What Can the Mitochondrial Disease Friedreich
- 1. School of Medicine, University of New Mexico, USA
- 2. Department of Internal Medicine, University of New Mexico, USA
Abstract
Friedrich’s Ataxia (FA) is a progressive neurodegenerative disorder commonly cited with a incidence of 1/50,000 live births. The disease is autosomal recessive and arises from a trinucleotide GAA repeat on chromosome 9. This repeat is located within the frataxin gene, which is located within the mitochondria and is commonly cited to be closely related to iron homeostasis within the mitochondria. Thus, FA is a neurodegenerative disease primarily arising from mitochondrial dysfunction. Clinically, FA presents in the first two decades of life, and is associated with significant morbidity and mortality. Clinical findings include truncal and limb ataxia, and absent deep tendon reflexes. Other findings include pes cavus, scoliosis, cardiomyopathy and dysarthria. FA results in several morphological, functional and biochemical abnormalities on the cellular and mitochondrial level. Moreover, several mitochondrial alterations have been linked to aging and age related diseases. The present mini-review will compare the common abnormalities found in the mitochondria due to normal aging and FA.
Keywords
Friedreich ataxia, Aging, Mitochondria
CITATION
Delahoussaye M, Raizada V. What Can the Mitochondrial Disease Friedreich’s Ataxia Tell Us about the Aging Mitochondria? J Cardiol Clin Res. 2023; 11(2): 1190.
INTRODUCTION
Friedrich’s Ataxia is a neurodegenerative ataxia, afflicting anywhere between 1/15,000 to 1/200,000 of the population with geographical variation [1]. Patients with Friedrich’s Ataxia often present with ataxia, diabetes mellitus, and cardiac disease. The disease commonly presents in the first two decades of life with truncal and limb ataxia and absent deep tendon reflexes being among the most consistent clinical findings within 5 years of diagnosis. Other clinical findings include dysarthria (difficulty speaking), loss of joint position and vibratory sense, scoliosis, pes cavus, and cardiomyopathy. Indeed, most patients with FA will develop cardiomyopathy at some point in their life. Most patients are wheelchair bound within 15 years of diagnosis, or by age 25 [2].
The disease is caused by a mutation in the Frataxin gene which was first identified in 1996 [3].The genetic alterations occur due to a GAA tri-nucleotide repeat on chromosome 9 in the frataxin gene. Expansion of these repeats results in a misfolded and/or non-functioning frataxin protein, and decreased number of frataxin transcripts [4]. Normally, the protein contains less than 50 of these GAA repeats, whereas these expansions in Friedrichs ataxia may contain hundreds of trinucleotide repeats. The disease severity and age of onset is positively associated with the number of GAA repeats, with the age of onset occurring 2-3 years earlier for each additional 100 GAA repeats. Moreover, the number of GAA repeats is also correlated with a more rapidly progressing disease course [5].
Normal Mitochondrial Function and Morphology
Mitochondria are cellular organelles that are essential to eukaryotic life. Interestingly, mitochondria carry their own DNA, separate from the DNA which resides in the nucleus of the cell proper. This DNA is maternally inherited. They perform a variety of functions, including cellular respiration, fatty acid oxidation, ATP production, iron regulation and heme synthesis, among others [6]. Moreover, mitochondrial morphology has been studied extensively in regard to healthy mitochondria, as well as diseased and aging mitochondria. Healthy mitochondria have been shown to form networked arrays. This is thought to contribute to efficient energy production as well as aid in fission and fusion [7]. Individual mitochondria also have characteristic morphology, which can vary between which tissue the mitochondria reside in. Typically, they are represented as oblong structures, with an inner and outer membrane, with the inner membrane folding over on itself to form cristae [8]. It has been documented that diseased or stressed mitochondria can lose their characteristic morphology and network. These mitochondrial may decreased in size, or lose their shape or cristae [9] (Figure 1).
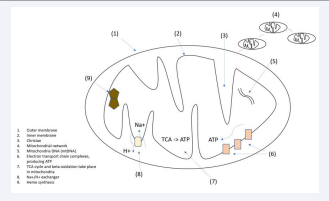
Figure 1: Normal Mitochondrial Function and Morphology.
The Aging Mitochondria
Mitochondrial changes and dysfunction have long been linked to aging. Many changes to mitochondrial function and morphology have been shown to occur with aging over the past several decades. However, whether these changes are a byproduct of aging or whether they contribute to aging directly remains to be conclusively established.
In muscle tissue, mitochondrial ATP production and mitochondrial VO2 max both decreased by 5-8% per decade, when accounting for covariates. Moreover, mitochondrial protein expression of 12 proteins of interest declined by 8-10% per decade of life. Finally, and importantly, mtDNA content decreased with age and DNA oxidation increased with age [10]. What causes this oxidative DNA damage? Many have postulated that oxidative damage from free radicals was the primary driver of oxidative damage to mitochondria [11]. Several studies have been published which both support and refute this claim.
With ageing, mitochondria tend to exhibit decreased membrane potential and increased ROS production [12]. They also can develop and accumulate lipid particles, as seen in hepatic steatosis [13]. Non-heme iron has been shown to accumulate in mitochondria with age. This correlated with mtRNA damage and Ca++ handling capacity [14]. Non-heme iron has also been shown to accumulate in the cytosol and mitochondria in the brain cortex [15]. Finally, in C.elegens, mitochondria tend to fragment, have decreased volume, and a decrease in network capacity [16] (Figure 2).
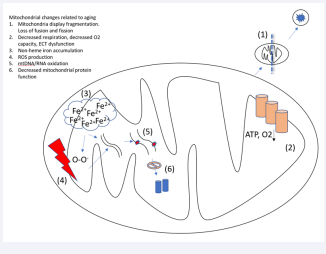
Figure 2: Mitochondrial changes related to aging.
Mitochondrial Pathophysiology in Friedrich’s Ataxia
Frataxin is a protein that is primarily found in the mitochondria, and has been located on normal human tissue in the muscle, heart cerebellum and spinal cord among other tissues [3]. Indeed, a review article from 2019 touched on the role frataxin deficiency in FA has been shown to contribute to deficiency in Fe-S clusters, iron overload in the mitochondria, and oxidative stress, and inflammation. Frataxin has many functions, including participating in the formation of essential Iron-sulfur clusters in the mitochondria [17,18]. These clusters have an important role in mitochondrial ECT function and respiration. It has been shown that a deficiency in any one of the proteins responsible for formation of the Fe-S clusters results in an accumulation of ‘free’ iron not related to heme or Fe-S synthesis [19]. Moreover, the dysfunction of frataxin and Fe-S clusters has been demonstrated to be linked to iron overload in the cytosol and mitochondria of cardiac myocytes in patients with FA. About 1-10% of cardiac myocytes will have iron granules, revealing a multifocal pattern of distribution [20].
In vitro frataxin knockdown cells were shown to result in mitochondrial dysfunction, altered mitochondrial membrane potential, reduced mitochondrial respiration and overall ATP production, and reduced cofactors essential for the formation of heme. In Frataxin knockdown cells, the mitochondria appeared fragmented and localized around the nucleus in juxtaposition to controls which were networked. Cells without frataxin exhibited hyperpolarized mitochondrial membrane potential which has been linked to ROS production [21]. As a consequence of increased iron accumulation and production on ROS, FA model mice have shown to lead to increased levels of neighboring lipid peroxidation in the mitochondria. Production of these lipoperoxidation products contributes to damage of cellular compartments and eventual cell death [22].
Cells without frataxin also had decreased cellular respiration, O2 utilization, and decreased ATP production. In addition, cells that were rescued with an iron storage protein in frataxin suppressed cells could reduce the accumulation of free iron, returning some normal mitochondrial function. Finally, the authors showed that frataxin knockdown resulted in higher rates of ferroptosis compared to controls [23,24].
In Mice with frataxin knock out, the mitochondria exhibited structural abnormalities such as decrease in number and with increased size and irregular cristae. These mitochondria also had lower rate of oxygen consumption [25] (Figure 3).
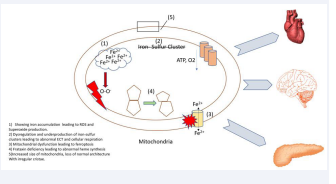
Figure 3: Mitochondria showing iron accumulation leading to ROS and Superoxide Production.
Iron, Diabetes in FA
Many patients with FA go on to develop type 2 diabetes, with estimates reported between 10%-30% of patients [26]. A large multicenter study published in 2022 found an 8.7% incidence of DM in FA patients [27]. Research has shown that individuals with T2D have decreased levels of leukocyte frataxin compared to controls without T2D. This association was independent of confounding factors such as gender, albumin, hypertension and BMI. Notably, these patients with T2D were not patients with FA but still had a relative deficiency in frataxin [23]. These findings add to the growing body of evidence that mitochondrial dysfunction has a significant role in the development of insulin resistance and type 2 diabetes. It has been shown previously that acquired defects in mitochondrial proteins can directly cause Insulin resistance, the hallmark of T2D [29].
Frataxin knockout mice were shown to have decreased islet cell mass and islet cell number, leading to decreased insulin secretion capacity. Notably, the islet cell number was identical in both young FA knockout mice and young controls. However, the FA knockout mice showed reduced islet cell mass, suggesting the loss of mass was acquired as the mouse aged. Frataxin deficiency also leads to an increase in ROS and free radicals in pancreatic islet cells, predominately superoxide. The increase in ROS is thought to be related to increased free iron in the cell and mitochondria secondary to frataxin deficiency. Dysregulated iron has been linked to an increase in mitochondrial ROS [30].
Mitochondria, Cardiomyopathy in FA
Cardiomyopathy is one of the primary derangements seen in FA. It is seen in up to 1/3 of patients and is the single leading cause of death in FA [3]. Both lipofuscin and free iron aggregates have been found in the cardiomyocytes of FA patients, along with the typical fibrosis and hypertrophy expected from cardiomyopathy [31]. One study showed that Frataxin KO mice exhibited a relative cardiomyocyte cytosolic iron deficiency, along with mitochondrial iron overload. Moreover, they showed that iron supplementation could limit the extend of cardiomyopathy, presumably by rescuing cytosolic iron content. They also postulated that the relative Frataxin deficiency in certain organs led to alterations in systemic iron homeostasis [32]. However, there is debate as to if iron accumulation is a causative factor in organ dysfunction or a incidental byproduct. Although the toxic effects of frataxin loss have been documented on a cellular level, some studies have shown that frataxin deficiency without iron overload and sufficient to cause embryonic death. This is consistent with the model of human FA, in which some frataxin expression is preserved, at least for some time [2,33].
DISCUSSION
Friedrich’s Ataxia is a mitochondrial disease leading to significant morbidity and mortality in this afflicted. Research has elucidated several morphological, functional, and biochemical abnormalities seen in the mitochondria of patients with FA or models with an equivalent frataxin deficiency [17,18,25]. Moreover, the mitochondrion have become an area of interest and research in regards to the role they potentially play in aging. Indeed, both aging mitochondrion and those in FA have been shown to exhibit impaired iron homeostasis and accumulated of non-heme iron [14,31]. In FA, this accumulation is thought to be due to a relative deficiency in Frataxin, and occurs at a highly accelerated rate compared to non-diseased cells. In a normal aging cell, this accumulation occurs over many more decades and is thought to be due to cumulative effects of inflammation and ROS on mitochondrial proteins and mtDNA.
Moreover, OXPHOS and respiratory dysfunction is seen in both FA cells and in aging cells. Aging mitochondria in muscle cells shows a 5-8% decrease in ATP production and VO2max. Similarly, FA cells in vitro had a significant decrease in ATP production and O2 consumption. The decrease in the frataxin model was identified as being due to a defect in complex IV of the ECT. In aging cells, the decrease was postulated to be due to an accumulation of ROS, mtDNA damage, and a decrease in protein function and number [10,24].
Morphological changes occur in both FA, frataxin deficient models, and in normal aging. In FA models, mitochondria exhibited a decrease in number, size, and abnormalities in cristae structure [25]. Mitochondria in C.elegens exhibit fragmentation, decreased volume, and a decreased capacity to form mitochondrial networks.
CONCLUSION
Mitochondrial function is significantly altered in FA and FA models, and in normal aging. Several alterations are common to both, such as irregular iron homeostasis, decreased oxidative capacity, increased production of ROS, mtDNA damage, and morphological changes. Despite these abnormalities, it is unclear as to which abnormalities are a consequence of aging, or a direct driver of aging and dysfunction.
Authors Contributions
Michael Delahoussaye, School of Medicine, University of New Mexico.
Veena Raizada, Department of Internal Medicine, Cardiology Section, University of New Mexico, Albuquerque, NM 87106, United States.









































































































































{kind=link}