Identification of Atypical Pml or Rara Fusion Transcript by Molecular, Cytogenetics and Flow Cytometry Analysis: A Case Report
- 1. Hematology and Transplantation CSE, Cardarelli Hospital, Italy
- 2. Molecular Biology Laboratory, Hematology and Transplantation CSE, Cardarelli Hospital, Italy
- 3. Unit of Transfusional Medicine, Cardarelli Hospital, Italy
- 4. Molecular Biology and Clinical Genetic Unit, Cardarelli Hospital, Italy
- 5. Department of Molecular Medicine and Biotechnology, University Federico II
- 6. Ceinge- Advanced Biotechnologies Center, Italy
- 7. Stem Cell Transplant Program, AORN Cardarelli, Naples
- 8. Department of Biomedicine and Preventive, Tor Vergata University, Italy
Abstract
Acute Promyelocytic Leukemia (APL) is characterized by the PML-RARA fusion gene, as a consequence of the t (15;17(q22;q21) translocation. Depending on the PML breakpoint, usually located within intron6, exon6, or intron 3, different PML/RARA transcript isoforms may be generated: long (bcr1), variant (bcr2), and short (bcr3), respectively. We report the characterization of a case of APL with atypical PML/RARA transcript, which was not clearly detectable using standard molecular procedure. Differential detection of PML-RARA bcr1, bcr2, bcr3 fusion transcripts were analyzed using a LAMP technology based kit (DiaSorin Molecular) and no amplification detected. Subsequently a Real time PCR performed using HemaVision-28Q kit (DNA Diagnostic), no detecting amplification. Given the strong clinical and morphological suspicion, we tested the sample for RT-PCR (Biomed protocol) using PML-A2 and RARA-B primers. Analysis of PCR product showed one specific band of 440 bp, a size that does not correspond with the recognized size of the typical transcripts.
The immunophenotipic characterization carried out on FACS CANTO II BD, showed 90 % of abnormal mononuclear cells 45% of which expressed CD34 and a myeloid phenotype (positive for CD117, CD13, CD33 and partial for CD2 and MPO). Apart from this population with the help of logical gate, we found a population that did not express CD34 and HLA DR and had cytometric characteristics of APL.
Analysis of the bone marrow (BM) aspirate demonstrated a markedly hypercellular marrow with 40% myeloblasts and 30% abnormal promyelocytes of medium-sized, with bilobed nuclei and hypergranulated cytoplasm and rare Auer bodies. The traditional cytogenetic analysis at the diagnosis, assayed by the R- banding, revealed the following karyotype: 46, XY, t(15;16;17) (q24;p13;q21) [18]/47, XY, +8, t(15;16;17) (q24;p13;q21)[2].
In this report, we describe a patient with this specific translocation involving three chromosomes: 15, 17 and 16.
Keywords
APL, atypical PML/RARA , Real Time PCR
ABBREVIATIONS
APL: Acute Promyelocytic Leukemia
Cite this article
Viola A, Muggianu SM, Peluso R, Caputo M, Pisa M, et al. (2022) Identification of Atypical Pml/Rara Fusion Transcript by Molecular, Cytogenetics and Flow Cytometry Analysis: A Case Report. J Hematol Transfus 9(1): 1106
INTRODUCTION
Acute Promyelocytic Leukemia (APL) is a subtype of Acute Myeloid Leukemia (AML) with specific clinical and biological features accounting for about 10 percent of cases [1].
The disease is characterized by the PML-RARA fusion gene, as a consequence of the t(15;17(q22;q21)translocation. Depending on the PML breakpoint, usually located within intron6, exon6, or intron 3, respectively, different PML/RARA transcript isoforms may be generated: long (bcr1), variant (bcr2), and short (bcr3), respectively. These fusion genes are sensitive to retinoid differentiating agents, such as all-trans retinoic acid (ATRA) and antiapoptotic agent arsenic trioxide [2].
Although t (15;17) (q22; q21) has been detected in ~90% of APL patients, variant translocations have been reported in a few APL patients; in particular, chromosomal rearrangements in addition to t (15;17) occur 25-40% of APL patients with a large preponderance of trisomy 8.
We report the characterization of a case of APL with atypical PML/RARA transcript, which was not clearly detectable using standard molecular procedure needing additional investigation for the identification of the PML-RARA fusion.
CASE PRESENTATION
A 31year old man presented at the Emergency and then to Hematology Unit of Cardarelli Hospital (Naples) in April 2021 with malaise, intermittent vomiting and an asthenia. A blood examination demonstrated a hemoglobin (Hb) count of 130 g/l, a white blood cell (WBC) count of 5.02x109/l, with 50 % atypical cells and a platelet count of 28x109/l.
Coagulation tests identified a prothrombin time of 1.82, a normal prothrombin international ratio of 0.86, a fibrinogen level of 100 mg/l and a D-dimer of 2955 ng/ml. Analysis of the bone marrow (BM) aspirate demonstrated a markedly hypercellular marrow with 40% blasts, characterized by medium to large cells with altered nucleus/ cytoplasm ratio, and 30% abnormal promyelocytes, of medium-sized, with bilobed nuclei and hyper granulated cytoplasm and rare Auer bodies (Figure 1).
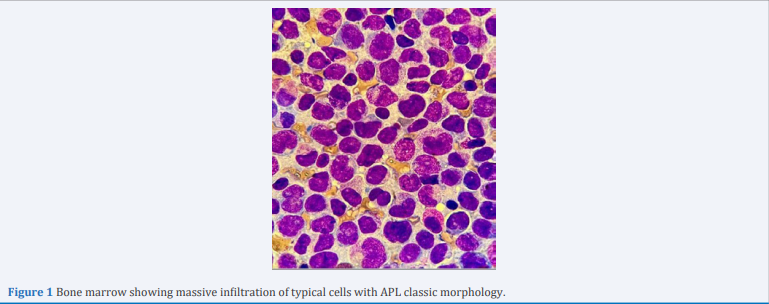
Figure 1 Bone marrow showing massive infiltration of typical cells with APL classic morphology
The immunophenotipic characterization carried out on FACS CANTO II BD, showed 90 % of abnormal mononuclear cells 45% of which expressed CD34 and a myeloid phenotype (positive for CD117, CD13, CD33 and partial for CD2 and MPO). Apart from this population with the help of logical gate, we found a population that did not express CD34 and HLA DR and had cytometric characteristics of APL (Figure 2).
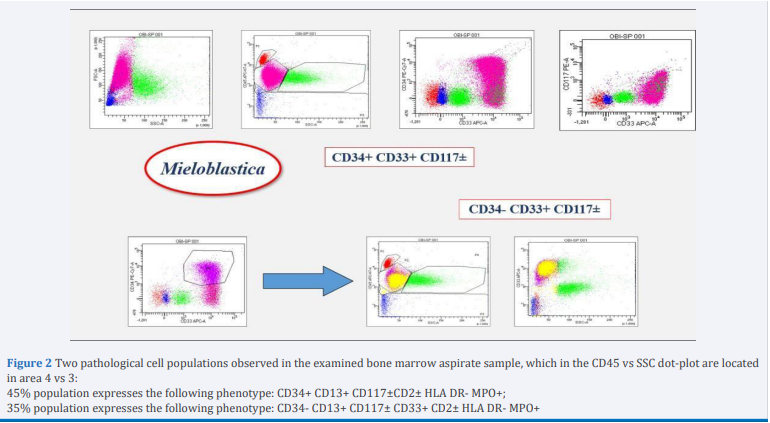
Figure 2 Two pathological cell populations observed in the examined bone marrow aspirate sample, which in the CD45 vs SSC dot-plot are located in area 4 vs 3:
45% population expresses the following phenotype: CD34+ CD13+ CD117±CD2± HLA DR- MPO+;
35% population expresses the following phenotype: CD34- CD13+ CD117± CD33+ CD2± HLA DR- MPO+
Reverse transcriptase-polymerase chain reaction (RT-PCR) performed differential detection of PML-RARA bcr1, bcr2, bcr3 fusion transcripts were analyzed using a LAMP technologybased kit (Dia Sorin Molecular) and no amplification detected. Subsequently a Real Time PCR performed using HemaVision28Q kit (DNA Diagnostic), no detecting amplification. Given the strong clinical and morphological suspicion, we tested the sample for RT-PCR (Biomed protocol) using PML-A2 and RARA-B primers and PML-A1 and RARA B. Analysis of PCR product using first two primers showed one specific band of 440 bp, a size that does not correspond with the recognized size of the typical bcr3 transcripts (Figure 3A), while using the other primers that identify bcr-1 transcript no amplification was detected (Figure 3B).
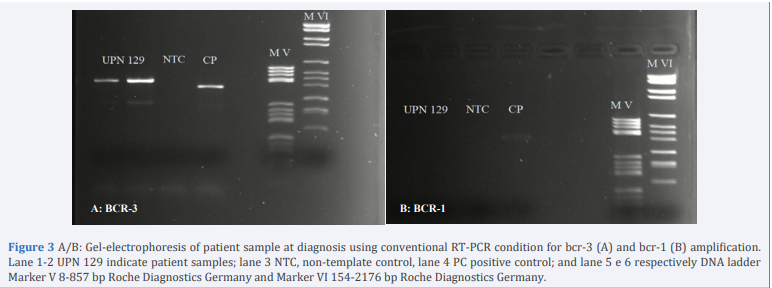
Figure 3 A/B: Gel-electrophoresis of patient sample at diagnosis using conventional RT-PCR condition for bcr-3 (A) and bcr-1 (B) amplification. Lane 1-2 UPN 129 indicate patient samples; lane 3 NTC, non-template control, lane 4 PC positive control; and lane 5 e 6 respectively DNA ladder Marker V 8-857 bp Roche Diagnostics Germany and Marker VI 154-2176 bp Roche Diagnostics Germany.
The traditional cytogenetic analysis at the diagnosis, assayed by the R- banding, revealed the following karyotype: 46, XY, t(15;16;17) (q24;p13;q21)[18]/47,XY,+8,t(15;16;17) (q24;p13;q21) (Figure 4).
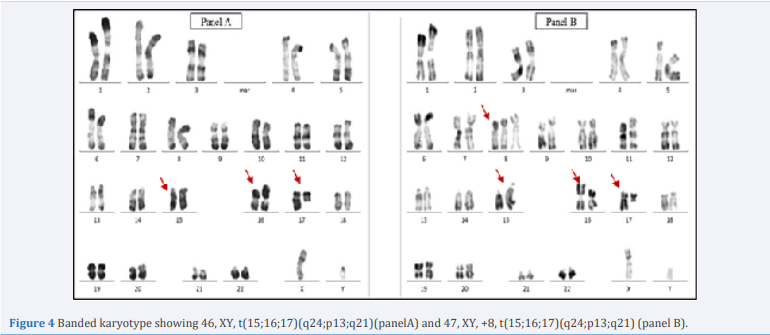
Figure 4 Banded karyotype showing 46, XY, t(15;16;17)(q24;p13;q21)(panelA) and 47, XY, +8, t(15;16;17)(q24;p13;q21) (panel B).
This report describes a patient with this specific translocation involving three chromosome: 15, 17 and 16. Another studies reported a case with the coexistence of t(15;17) and complex variant t(15;16;17)(q22;q24;q21) [3]. In order to get a correct identification of transcript so to perform sensitive and specific quantitative evaluation of MDR during the follow up we carried out direct sequencing of PCR product.
The patient was diagnosed as APL according to the results of bone marrow morphology, cytogenetics, and molecular biology, with a rare PML RARA point of fusion. The patient was started on all-trans retinoic acid (ATRA) at 45?mg/m2 orally on days 1 to 28 (4 tablets h 7.00 am and pm / daily), arsenic trioxide (ATO) (10 mg ev / daily) intravenously on days 3 to 28 (from day +8 until day+17 he underwent a reduction to half a dose because of ponderal increase). At the same time, frequent plasma and blood transfusion, infection control, and other symptomatic support treatment given to the patient. On May 19, 2021, he was discharged at home; the blood routine showed a leukocyte count of 3.37?×?109 /L N 51 %, a hemoglobin level of 10.1 g / dl, and a platelet count of 261?×?109 /L. Subsequently he underwent consolidation therapy (ATRA+ATO) for three cycles, according to literature [4-6], keeping molecular complete remission. Actually, the patient is in good health conditions.
DISCUSSION
In the present report, we described a case of patient with APL characterized by novel atypical PML/RARA fusion transcript. Result of standard molecular and immunophenotipic analysis was not clear while the morphology and the clinical features were highly suggestive for APL.
In most cases the best diagnostic option for APL is RT-PCR testing; however, each testing modality has its own advantages.
Conventional karyotype can identify the usual t(15;17) and may also identify other genetic abnormalities that have prognostic significance, but requires longer response time and this is not acceptable in APL.
RT-PCR is the most sensitive test, also used for monitoring MRD as well as to identify cryptic translocation, and allows the identification of the specific translocations subtype, including bcr- 1,-2,-3.
In our case, using different molecular test such as Lamp technologies based Kit (DiaSorin Molecular) and a Real time PCR using HemaVision-28Q kit (DNA Diagnostic) no amplification detected.
Then RT-PCR for PML/RARA detection was carried out using standard protocol(Biomed) and the analysis of sample by gel – electrophoresis showed an atypical size respect classic size of the typical isoform (bcr1,bcr2,bcr3).
In this case, it was necessary to carry out additional investigations for the identification of the PML-RARA isoform.
For this reason, we used the Sanger sequencing approach, a methodology used to study gene regions associated with a defined phenotype, to confirm next generation sequencing (NGS) variants, to detect minor allele fractions up to 5%, or read contiguous sequences up to 1,000 bases. Gene fragment analysis based on simple and straightforward workflows and is used in a wide range of applications, such as mutation detection, genotyping, identification of short tandem repeats, and gene expression profiling.
The methodology used in this case report confirmed the sequence identified through the cytogenetic approach; in addition, it allowed establishing that the PML-RARA fusion gene sequence had kept the reading code unchanged.
The final diagnosis made was APL and the patient started therapy with arsenic trioxide and all- trans-retinoic-acid. Nowadays he is in complete remission after 18 months after diagnosis. Actually, to the best of our knowledge, 52 cases with complex translocation have been previously reported [7-10].
The majority of previous studies observed no difference in clinical outcome between APL with typical t(15;17) and APL with complex translocations [11,12].
This case indicates that the use of different experimental approaches and different techniques is necessary to confirm the diagnosis of difficult cases of APL and is fundamental for minimal residual disease (MRD) monitoring.









































































































































{kind=link}