OMICS of Beta Thalassemia: Review Article
- 1. Department of Clinical Haematology, National Institute of Blood Diseases and Bone Marrow Transplantation
- 2. Department of Research & Development, National Institute of Blood Diseases and Bone Marrow Transplantation
Abstract
Background: Beta - Thalassemia is the most common hemoglobinopathy throughout the world. Beta globin chain synthesis is completely or partially compromised in the affected individuals. This leads to transfusion dependency in majority of patients. However, despite of genetically proven beta thalassemia major transfusion requirement in some patients is negligible or seldom required.
Introduction: Around 400 mutations have been identified till now, among which five to six are common in a certain region. For example IVSI-5(G-C) is commonest mutation in the subcontinent. These mutations act as the primary modifiers. For example mutations in the promoter region lead to less severe disease phenotype while on the other hand, deletions or frame shifts result in severe phenotype. Similarly there are secondary genetic modifiers that are polymorphisms that can modify the phenotype. Xmn1 polymorphism, BCL 11 gene polymorphisms, co inheritance of alpha thalassemia are the well-studied examples. These polymorphisms are associated with increased Hb F especially under stressful conditions like pregnancy, severe anemia, etc. Hb F is the major hemoglobin in the fetal life due to hypomethylated Cp G islands of beta and gamma genes while they become methylated in adult life leading to switching of Hb class. This was the basis of trials of different Hb F augmenting agents. Different studies have shown that individuals harboring certain (IVSI-1, IVSI-5, CD 30, CD 15, etc) primary and secondary modifiers (Xmn1 polymorphism, BCL polymorphism) show promising results with Hb F augmenting agents. These genetic factors can help to formulate the management of thalassemia patients without or with minimum blood transfusions.
Conclusion: Genetic studies of beta Thalassemia patients should be performed as it can help in planning the long term management of these patients. Patients harboring certain primary and secondary genetic modifiers can be managed without blood transfusion.
Keywords
Beta thalassemia; Xmn1 polymorphism; Hemoglobinopathy; Hb F augmenting agents
Citation
Siddiqui S, Jamal A, Sheikh A, Shamsi T (2022) OMICS of Beta Thalassemia: Review Article. J Hematol Transfus 9(1): 1102.
ABBREVIATIONS
HBB: Beta globin gene, UTRs: Untranslated region, LCR: Beta Locus control region, P: Promoter region, HU: Hydroxyurea
INTRODUCTION AND EPIDEMIOLOGY
Beta thalassemia, an autosomal recessive disorder, is the most common hemoglobinopathy throughout the world [1]. It is characterized by reduced or completely absent beta globin chain synthesis [2]. Around 1.5% population of the world is carrier for this disease [3], while carrier rate in our country is around 5%. Historically beta thalassemia was highly prevalent in Mediterranean region, Southeast Asia and Middle East. However, with increasing trend of migration the prevalence of beta thalassemia is increasing in different other regions of the world like Northern Europe and America [4]. On the contrary, the incidence of this disease has markedly reduced in certain countries like Italy and Cyprus, due to stringent prevention and treatment strategies [5]. Still certain regions like South East Asia, due to political and financial factors, are struggling against the high prevalence of this disease.
Molecular Epidemiology
Globally there are around 400 different mutations in beta globin gene [6]. Some of them lead to severe transfusion dependent phenotype (β0) as there is no production of beta chains at all, while on the other hand, some mutations are β+ that result in milder phenotype or Thalassemia intermedia [7]. Majority of the beta thalassemia mutations are point mutations. Among the 394 mutations detected till now, only 5-6 mutations are common and specific for any region/ ethnicity. For example in Pakistan the commonest mutations are IVSI-5, Frame shift 8-9, Del 619, IVSI-1, Frame shift 41-42, and Codon 30. These above mentioned mutations are shared among different countries of Southeast Asia especially Pakistan, Iran and India, mainly due to mass migration of people during partition of India and Pakistan in 1947. Codon 15 and Codon 5 are relatively less common in our population [8]. Black et al have described the distribution and sharing of different genetic mutations among different parts of Pakistan, India, Srilanka, Oman and UAE [9].If we compare the genetic profiles of these countries we can see similarities in these profiles. For example IVS-1-5 (G–C), Fr 8/9 (+G), Fr 41/42 (–TTCT), IVS-1-1 (G–T) and Del 619 are prevalent in Pakistan and India. IVSI 5(G-C) is common throughout the subcontinent and reported as highly prevalent in indigenous Indian tribes [10]. In Pakistan 80% of Baloch community suffering from beta thalassemia inherits this mutation pointing towards possibility of origin of this mutation in this region. This mutation is shared by the residents of southern Iran and extends to Balochs of Oman and UAE whose ancestors moved there for trading in 18th century [11-12]. Similarly, Del 619 is common in Gujrati population of both the countries. This mutation is common among Pakistani Sindhi and Memons while being also prevalent in Lohana community of India. It should be noted that historically Memons were called Lohana [8]. On the other hand if we examine the thalassemia genetic profile of Arabs, Europeans and Americans, it is completely different [13].
Genetic factors as modulators of beta thalassemia
Phenotypic ally, Beta thalassemia is a very heterogeneous entity. Severity of the disease ranges from lifelong transfusions starting from 6 months of life in beta thalassemia major to totally asymptomatic thalassemia minor individuals. The third entity is of Thalassemia intermedia, those individuals who are genetically thalassemia major but seldom require blood transfusions and maintain a good quality of life with Hb around 7-8g%. The phenotype of beta thalassemia is regulated by primary, secondary and tertiary genetic modifiers [14]. In this review we will be focusing primary and secondary genetic modifiers only, as role of tertiary genetic modifiers is beyond the scope of this article.
Primary genetic Modifiers
The production of beta globin chains is controlled by the Beta globin gene (HBB). It is located on chromosome 11 and spans 1.6 Kb. It is comprised of 3 exons along with 5’ and 3’ ends and the (UTRs) un translated regions. These exons and UTRs are regulated by the promoter region containing the TATA, CAAT and duplicated CACCC boxes. Expression of beta gene is also regulated by (LCR) locus control region that is 50 Kb from HBB gene. The HBB gene, LCR and UTRs together are known as primary genetic modifiers that are shown in (Figure 1).
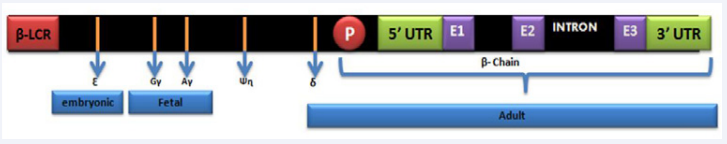
Figure 1 Diagrammatic representation of Chromosome 11 with details of HBB gene: E1: exon 1, E2: exon 2, E3: exon3; β LCR: Beta Locus control region; UTR: Untranslated region; P: Promoter region.
Following are the four major mutants: promoter mutant, RNA splicing mutant, RNA capping/tailing mutant, and a translation mutant. In addition to these, frame shift mutation is another defect, that is a nonsense mutation and results in premature termination of protein synthesis and hence, affecting globin synthesis [15]. It is now a well-known fact that these different mutations produce variable phenotypic effect as shown in (Table 1) [16-17].
|
Table 1: Common types and severity of Beta Thalassemia mutations in different countries of the world [58]. |
|||
|
S.No |
Countries |
Common mutations |
Severity |
|
1 |
Pakistan |
IVSI-5(G-C) IVSI-1(G-T) Fr8-9 Fr41-42(-TCTT) Del 619bp |
β+ β+ β0 β0 β0 |
|
2 |
India |
IVSI-5(G-T) Del 619 bp Fr 8-9 IVSI-1(G-T) |
β+ β0 β0 β+ |
|
3 |
Iran |
IVSI-5(G-C) IVSI-1-110(G-A) CD 5-CT Fr 8-9 |
β+ β+ β0 β0 |
|
4 |
Indonesia |
IVSI-5(G-T) IVSI-II-654(C-T) IVSI-1(G-T) -28 Cd15(G-A) |
β+ β0 β0 - β0 |
|
5 |
China |
Fr 41-42 (-TCTT) Cd 17 (A-T) IVSI-II-654(C-T) |
β+ β0 β0 |
|
6 |
Saudi Arabia |
Cd 39 IVS11-1(G-A) IVSI I-110(G-A) IVSI-5 (G-C) FR 8-9 IVSI-3’ end 25bp |
β0 β0 β+ β+ β0 β0 |
|
7 |
Greece |
IVSI-110(G-A) Cd 39(C-T) IVSI-1(G-A) IVSI-6(C-T) |
β+ β0 β+ β+ |
|
8 |
Thailand |
Fr 41-42(-TCTT) Cd 17(A-T) IVSI-5(G-C) IVSI-1(G-T) Cd 71-72(+A) |
β0 β0 β+ β+ β0 |
|
9 |
Italy |
Cd 39(C-T) IVSI-1(G-A) IVSI-110(G-A) |
β0 β+ β+ |
|
10 |
Turkey |
IVSI-110(G-A) IVSI-6(C-T) IVSII-1(G-A) |
β+ β+ β+ |
|
Abbreviations: IVSI-5(G-C): Intervening Sequence I-5; IVSI-1(G-T): Intervening Sequence I- 1; Fr8-9: Frame shift 8-9; Fr41-42(-TCTT): Frame shift 41-42; Del 619bp: Deletion 619 base pair; IVSI-1-110(G-A): Intervening Sequence; CD 5-CT: Codon-5; IVSI-II-654(C-T): Intervening Sequence; IVSI- 1(G-T): Intervening Sequence; Cd15(G-A): Codon- 15; Cd 17 (A-T): Codon- 17; Cd 39: Codon-39; IVSI-1(G-A): Intervening Sequence; IVSI-6(C-T): Intervening Sequence; Cd 71-72(+A): Codon- 71-72 |
|||
For example; if mutation occurs in promoter region or Poly A cleavage site it results in diminished synthesis of beta globin chains leading to β+ thalassemia which is usually less severe form of disease. These patients usually present later in life or need blood transfusions occasionally and maintain a good quality of life at Hb of 7-8g%. Following are the few examples of the promoter or UTRs mutations: -88 C>T, -88 C>A,-90C>T, -31 A>G and -30 T>A. On the other hand, β0 thalassemia occurs due to splice site mutation, frame shifts, deletion and initiation codon mutation as these lead to complete absence of beta chains and hence, severe transfusion dependent anemia [2, 18-19]. Del 619, Fr 8-9 and Fr 41-42 are few examples of β0 thalassemia [20].Genetic mutations in HBB gene can therefore modify the disease behavior and hence are primary genetic modifiers (Figure 2).
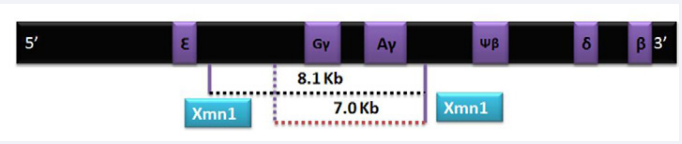
Figure 2 Chromosome 11 with both G? and A? genes.XMN 1 normally produces an 8.1 kb fragment while a polymorphic site due to C-T substitution at -158 5’to G? produces a shorter segment of 7 kb.
Secondary genetic modifiers
Because of the absence of beta chains there is relative excess of alpha chains that precipitate and lead to shortened red cell survival and increased ineffective erythropoiesis. The secondary genetic modifiers regulate the levels of α or ? globin gene expression in response to decreased levels of beta globin gene and therefore ameliorates the disease phenotype [21]. The well-known secondary factors include Xmn1 polymorphism, BCL11 and HBS1-MYB and co inheritance of α thalassemia [22- 23]. Various recent studies have revealed the co relation of these three modifiers with improved Hb F in adults [24].
Xmn1 polymorphism is a C-T substitution at position -158 of the Gγ globin gene [25]. Its frequency in different population is reported as 0.32-0.35 .Position of XMN 1 is close to LCR region of HBB gene which, as described above, controls HBB gene expression throughout life. It has been postulated that this polymorphism reduces the binding of transcription inhibitory factors to the LCR and hence reactivates the G? genes in adult life leading to increased production of ? globin chains that pair with excess alpha chains. This reduces the imbalance between α and non α chains leading to decreased ineffective erythropoiesis and increased production of Hb F. Therefore, the net result is increased or stable Hb. This polymorphism usually induces Hb F production under stressful conditions like severe anemia and pregnancy, and has no significant clinical effect in normal population [26-27]. Sharma et al have reported that patients of beta thalassemia major with homozygous or heterozygous Xmn 1 polymorphism usually have higher baseline hemoglobin level and present later [28]. This polymorphism is also associated with better response to different Hb F augmenting agents. Various studies from different part of the world have shown substantial role of Xmn1 polymorphism on ameliorating the disease severity [29-32]. Ansari et al reported that 69% of the patients harboring this polymorphism responded better to Hydroxyurea (requiring no transfusion or decreased blood transfusion) as compared to those who lacked this polymorphism [33].
BCL 11 polymorphism
Another well studied secondary genetic modifier is BCL 11 gene that resides on chromosome 2. It is a well-known suppressor of HbF. Several SNPs or variants in this gene that inhibit BCL 11 function augment Hb F. Uda et al and other studies have shown that C variant in rs11886868 is associated with increased Hb F and hence milder disease phenotype in homozygous beta thalassemia major patients [34]. A recent study has shown that loss of this region is associated with an increase in around 2.7% of Hb F that significantly affects the phenotype of beta Thalassemia major patients [35]. However, genome wide association studies have revealed that this polymorphism is associated with variable results in different populations and its role in Hb F augmentation is still controversial [36-37].
Coinheritance of Alpha Thalassemia
As described above excess of alpha chains is the major determinant of severity in beta thalassemia. Therefore, double heterozygous state of alpha/beta thalassemia leads to clinically less severe beta thalassemia .This has been reported from various regions of the world [38-39]. In Pakistan around 30% beta thalassemia major patients co inherit alpha thalassemia. Out of which 80% are single gene deletions, 19% two gene deletion and remaining triplicated alpha genotype. Most common type in our population is deletion of 3.7 kilo base pairs followed by 4.2 kilo base pairs [33, 40-41].
Role of Hb F augmenting agents in beta thalassemia
The non-alpha gene cluster i.e. β, ? and δ undergoes multiple changes during the process of ontogeny. Studies have shown that CpG islands in ? and β genes are hypomethylated during the fetal life. While in the adult life CpG islands of ? chains are methylated (1%). This was the basis on which DeSimone and colleagues conducted Hb F augmentation research in anemic baboons [42]. Later Hydroxyurea was shown to induce fetal hemoglobin production in vitro by inducing alterations in erythropoiesis. Because of its broader safety profile and oral route HU became the main drug as Hb F agent in hemoglobinopathies [43- 44]. However, exact and complete mechanism of HU as HbF augmenting agent is still not completely understood.
Difference in response to Hb F augmenting agents due to genetic mutations in different part of the world
Hydroxyurea has been extensively studied by different researchers throughout the world [44]. Initially, role of HU as Hb F augmenting agent in Beta Thalassemia major and intermedia remained controversial. However, subsequent clinical trials from India, Pakistan, Iran and other countries were very encouraging [45-48]. Reasons for variation in response among different ethnicities have been studied extensively.
Genetic mutations associated with better response
Ansari et al reported that patients carrying splice site or promoter mutations (e.g., IVSI-5 and IVSI-1) when treated with Hydroxyurea become transfusion independent or their transfusion requirement is reduced [50]. These patients maintain their Hb between 7-8 g %. On the other hand large deletions like Deletion of 619 base pairs and frame shifts like Fr 8-9 and Fr 41-42 are associated with absent production of beta chains and hence, results in severe transfusion dependent disease [49]. Genetic mutations in compound heterozygous state also ameliorate the disease phenotype e.g. IVSI-5 in homozygous state may reveal a beta thalassemia major phenotype while IVSI-5 with Cd 30 may reveal Beta Thalassemia Intermedia phenotype. This genotypic pattern can transform the management of Beta Thalassemia Major and Intermedia in Pakistan and its neighboring countries to a greater extent.
Effect of Xmn 1 polymorphism
Numerous studies have confirmed the positive effect of HU in XMN positive patients. This polymorphism has proven to be a strong modifier of disease even in those patients with β0 phenotype. Ansari and colleagues have shown that 38% patients with homozygous IVSI-5 but XMN negative were associated with thalassemia major phenotype while 92% patients harboring homozygous IVSI-5 along with XMN behaved as Thalassemia intermedia [50]. Similarly, Alebouyeh et al. showed HU to be effective in 69% transfusion-dependent patients. Majority of them were positive for this polymorphism [51]. Indian study conducted by Italia et al showed that 72.2% patients of Thalassemia intermedia who responded to HU were positive for XMN1 polymorphism [52].
Effect of BCL 11 polymorphism
Silencing of this gene is associated with less severe phenotype. Multiple ongoing or recent studies have shown variable response of HU in patients with this polymorphism. Banan et al from Iran studied the effect of XMM polymorphism and BCL 11 in patients receiving HU. They concluded a strong relationship of good response to HU in patients who were positive for XMN 1 and BCL 11 polymorphism [53]. While Sclafani and colleagues failed to establish any association with response to HU in 15 patients of Beta Thalassemia Intermedia [54]. Similarly Ansari et al also could not draw a statistically significant association between BCL 11 and response to HU [55].
Coinheritance of alpha thalassemia
Alpha thalassemia coinheritance in patients with beta thalassemia ameliorates the disease severity as unpaired excess alpha chains are less in number. This was proven by Panigrahi et al. who found α gene deletion to be an independent predictive factor for good response to HU therapy in patients with beta Thalassemia intermedia [56].
Combined effect of Genetic Factors
It can be postulated from different studies that response to HU is not dependent on a single genetic factor in majority of the cases. Different genetic combinations e.g. presence of IVSI-1 or Cd30 in homozygous or compound heterozygous state, along with presence of XMN1 polymorphism or co inheritance of alpha thalassemia can ameliorate the disease behavior and these genetic determinants can be utilized to foresee and plan the management of beta thalassemia patients [57].
CONCLUSIONS
Genetic studies of beta Thalassemia patients should be performed as it can help in planning the long term management of these patients. Patients harboring certain primary and secondary genetic modifiers can be managed without blood transfusion.
CONFLICT OF INTEREST
The authors declare that they have no competing interests
AUTHORS’ CONTRIBUTIONS
• Saima Siddiqui SS: Literature review ,drafting and intellectual input
• Aisha Jamal AJ: Critical review
• Affaf Sheikh AS: Literature review
• Tahir Shamsi TS: Basic concept
ACKNOWLEDGEMENTS
Safia Mehmood for graphics in manuscript
REFERENCES
- Weatherall DJ, Clegg JB. Historical perspectives: in The Thalassemia syndromes 4th edition.
- Origa R. Beta thalassemia. Genet Med. 2016
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization. 2008; 86: 480-487.
- Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders. Expert Rev Hematol. 2010; 3: 103-117.
- Angastiniotis MA, Hadjiminas MG. Prevention of thalassaemia in Cyprus. Lancet. 1981; 317: 369-371.
- Shamsi T, Hussain Z. An Insight into the Symptomatology of ß-Thalassaemia Major: Molecular Genetic Basis of the Disease–III. Nat J Hea Sci. 2018; 3: 1-2.
- Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013; 3: a011700.
- Ansari SH, Shamsi TS, Ashraf M, Farzana T, Bohray M, et al. Molecular epidemiology of β-thalassemia in Pakistan: Far reaching implications. Indian J Hum Genett. 2012; 18: 193.
- Black ML, Sinha S, Agarwal S, Colah R, Das R, Bellgard M, Bittles AH. A descriptive profile of β-thalassaemia mutations in India, Pakistan and Sri Lanka. J commun Genet. 2010; 1: 149-157.
- Colah R, Gorakshakar A, Nadkarni A, Phanasgaonkar S, Surve R, et al. Regional heterogeneity of β-thalassemia mutations in the multi ethnic Indian population. Blood Cells Mol Dis. 2009 ;42: 241-246.
- Eshghi P, Zadeh-Vakili A, Rashidi A, Miri-Moghadam E. An unusually frequent β-thalassemia mutation in an Iranian province. Hemoglobin. 2008; 32: 387-392.
- Amirian A, Karimipoor M, Jafarinejad M, Taghavi M, Kordafshari A, et al. First Report on the Co-inheritance of Beta-globin IVS-I-5 (GC) Thalassemia with Delta Globin CD12 {Asn-Lys (AAT-AAA)} HbA2 NYU in Iran. Arch Iran Med. 2011; 14: 8-11.
- Mashi A, Khogeer H, Abalkhail H, Khalil S. Molecular patterns of β-thalassemia mutations of Saudi patients referred to King Faisal Specialist Hospital and Research Center. J Appl Hematol. 2017; 8: 99.
- Weatherall DJ. Thalassemia as a global health problem: recent progress toward its control in the developing countries. Annals of the New York Academy of Sciences. 2010; 1202: 17-23.
- Galanello R, Perseu L, Satta S, Demartis FR, Campus S. Phenotype- genotype correlation in β-thalassemia. Thalass Rep. 2011; 1: e6.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010; 5:1-5.
- Danjou F, Anni F, Perseu L, Satta S, Dessì C, et al. Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. 2012; 97: 989.
- Rujito L, Sasongko TH. Genetic background of β thalassemia modifier: recent update. J Biomed Transl Res. 2018 ;4: 12-21.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010 ; 12: 61-76.
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Cambridge University Press; 2009.
- Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood. 2008; 112: 3927-3938.
- Guvenc B, Canataroglu A, Unsal C, Yildiz SM, Turhan FT, et al. β-Globin chain abnormalities with coexisting α-thalassemia mutations. Arch Med Sci. 2012; 8: 644.
- Mastropietro F, Modiano G, Cappabianca MP, Foglietta E, D’Asero C, et al. Factors regulating Hb F synthesis in thalassemic diseases. BMC Hematology. 2002; 2: 1-7.
- Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Gen. 2009; 18 (R2):R 216-223.
- Peri KG, Gagnon J, Gagnon C, Bard H. Association of-158 (C→ T)(Xmn I) DNA Polymorphism in G γ-Globin Promoter with Delayed Switchover from Fetal to Adult Hemoglobin Synthesis. Pediatr Res. 1997; 41: 214- 217.
- Garner C, Tatu T, Game L, Cardon LR, Spector TD, et al. A candidate gene study of F cell levels in sibling pairs using a joint linkage and association analysis. GeneScreen. 2000: 9-14.
- Mastropietro F, Modiano G, Cappabianca MP, Foglietta E, D’Asero C, et al. Factors regulating Hb F synthesis in thalassemic diseases. BMC Hematology. 2002; 2: 1-7.
- Sharma N, Das R, Kaur J, Ahluwalia J, Trehan A, et al. Evaluation of the genetic basis of phenotypic heterogeneity in north Indian patients with thalassemia major. Europ J Haematol. 2010; 84: 531-537.
- Miri-Moghaddam E, Bahrami S, Naderi M, Bazi A, Karimipoor M. Xmn1-158 γGvariant in B-thalassemia intermediate patients in South- East of Iran. Int J Hematol Oncol Stem Cell Res. 2017; 11: 165.
- Bandyopadhyay S, Roychowdhury K, Chandra S, Das M, Dasgupta UB. Variable severity of b-thalassaemia patients of eastern India: effect of a-thalassaemia and XmnI polymorphism. Clin Exp Med. 2001; 1: 155–159.
- Kaddah N, Rizk S, Kaddah AM, Salama K, Lotfy H. Study of PossibleGenetic Factors Determining the Clinical. J Med Sci. 2009; 9: 151-155.
- Peri KG, Gagnon J, Gagnon C, Bard H. Association of-158 (C→ T)(Xmn I) DNA Polymorphism in G γ-Globin Promoter with Delayed Switchover from Fetal to Adult Hemoglobin Synthesis. Pediatr Research. 1997;41: 214-217.
- Ansari SH, Shamsi TS, Munzir S, Khan MT, Erum S, et al. Gγ-Xmn I polymorphism: a significant determinant of β-thalassemia treatment without blood transfusion. J pediatr Hematol Oncol. 2013 ;35: e153- 156.
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, et al. Genome- wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Nat Acad Sci USA. 2008; 105: 1620-1605.
- Ghedira ES, Lecerf L, Faubert E, Costes B, Moradkhani K, et al. Estimation of the difference in HbF expression due to loss of the 5’δ-globin BCL11A binding region. Haematologica. 2013; 98: 305.
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, et al. Human fetal hemoglobin expression is regulated by the developmental stage- specific repressor BCL11A. Science. 2008; 322: 1839-1842.
- Galanello R, Dessi E, Melis MA, Addis M, Sanna MA, et al. Molecular analysis of beta zero-thalassemia intermedia in Sardinia. Blood. 1989; 74: 823-827.
- Al Qaddoumi AA. Co-inheritance of alpha and beta-thalassemia in a Jordanian family. Clin Lab Sci. 2006; 19: 165-168.
- Moghaddam ZK, Bayat N, Valaei A, Kordafshari A, Zarbakhsh B, et al. Co-inheritance of α-and β-thalassemia: challenges in prenatal diagnosis of thalassemia. Iran J Blood Canc. 2012; 2: 81-84.
- Shamsi T. Medical management of b-thalassaemia without blood transfusion: a myth or a reality? J Pak med Assoc. 2013; 63: 304-305.
- Saba Shahid MN, Zahid D, Hassan J, Ansari S, Shamsi T. Alpha thalassemia deletions found in suspected cases of beta thalassemia major in Pakistani population. Pak J Med Sci. 2017; 33: 411.
- DeSimone J, Heller P, Hall L, Zwiers D. 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Nat Acad Sci USA. 1982; 79: 4428-4431.
- Goren A, Simchen G, Fibach E, Szabo PE, Tanimoto K, et al. Fine tuning of globin gene expression by DNA methylation. PLoS One. 2006; 1: e46.
- Miller BA, Platt O, Hope S, Dover G, Nathan DG. Influence of hydroxyurea on fetal hemoglobin production in vitro. Blood. 1987; 70: 1824-1829.
- Algiraigri AH, Wright NA, Paolucci EO, Kassam A. Hydroxyurea for nontransfusion-dependent β-thalassemia: a systematic review and meta-analysis. Hematol Oncol Stem Cell Ther. 2017; 10: 116-125.
- Ansari SH, Shamsi TS, Ashraf M, Perveen K, Farzana T, Bohray M, Erum S, Mehboob T. Efficacy of hydroxyurea in providing transfusion independence in β-thalassemia. J Pediatr Hemato Oncol. 2011; 33: 339-343.
- Zamani F, Shakeri R, Eslami SM, RAZAVI SM, Basi A. Hydroxyurea therapy in 49 patients with major beta-thalassemia. Arch Iran Med. 2009; 12: 295-297.
- Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni AH, et al. Response to hydroxyurea in β thalassemia major and intermedia: experience in western India. Clin Chim Acta. 2009; 407: 10-15.
- Thein SL. Molecular basis of β thalassemia and potential therapeutic targets.Blood Cells Mol Dis. 2018; 70: 54-65.
- Ansari S, Rashid N, Hanifa A, Siddiqui S, Kaleem B, et al. Laboratory diagnosis for thalassemia intermedia: Are we there yet?. J Clin Lab Anal. 2019; 33: e22647.
- Alebouyeh M, Moussavi F, Haddad-Deylami H, Vossough P. Hydroxyurea in the treatment of major β-thalassemia and importance of genetic screening. Ann Hematol. 2004 ;83: 430-433.
- Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni AH, et al. Response to hydroxyurea in beta thalassemia major and intermedia: experience in western India. ClinChim Acta. 2009; 407: 10-15.
- Banan M, Bayat H, Azarkeivan A, Mohammadparast S, Kamali K, et al. The X mn I and BCL11A single nucleotide polymorphisms may help predict hydroxyurea response in Iranian β-thalassemia patients. Hemoglobin. 2012; 36: 371-380.
- Sclafani S, Pecoraro A, Agrigento V, Troia A, Di Maggio R, et al. Study on hydroxyurea response in hemoglobinopathies patients using genetic markers and liquid erythroid cultures. Hematology Rep. 2016; 8: 6678.
- Ansari SH, Hussain Z, Zohaib M, Parveen S, Kaleem B, et al. A Pragmatic Scoring Tool to Predict Hydroxyurea Response Among β-Thalassemia Major Patients in Pakistan. J Pediatr Hematol Oncol. 2022. 44: e77-e83.
- Panigrahi I, Dixit A, Arora S, Kabra M, Mahapatra M, et al. Do alpha deletions influence hydroxyurea response in thalassemia intermedia? Hematology. 2005; 10: 61-63.
- Panigrahi I, Agarwal S. Genetic determinants of phenotype in beta-thalassemia. Hematology. 2008; 13: 247-252.
- Boonyawat B, Monsereenusorn C, Traivaree C. Molecular analysis of beta-globin gene mutations among Thai beta-thalassemia children: results from a single center study. The Appli clin genet. 2014; 7: 253.









































































































































{kind=link}