Availability of DMD mRNA Transcripts Analysis in a patient with Dystrophinopathy having a Nonsense Mutation
- 1. Department of Medical Genetics, Shinshu University School of Medicine, Matsumoto, Nagano, Japan
- 2. Division of Rehabilitation, Nagano Children’s Hospital, Azumino, Nagano, Japan
- 3. Department of Neuromuscular Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry; Department of Clinical Development, Translational Medical Center, National Center of Neurology and Psychiatry, Kodaira, Tokyo, Japan
- 4. Department of Pediatrics, Shinshu University School of Medicine, Matsumoto, Nagano, Japan.
- 5. Department of Neurology and Rheumatology, Shinshu University School of Medicine, Matsumoto, Nagano, Japan
- 6. Intractable Disease Care Center, Shinshu University Hospital, Nagano, Japan
- 7. Department of Medical Genetics, Kitasato University Graduate School of Medical Sciences, Sagamihara, Japan
Keywords
• DMD
• BMD
• Dystrophin
• Exon-skipping
• RT-PCR
Citation
Narumi Y, Fueki N, Hayashi Y, Shiba N, Nishino I, et al. (2013) Availability of DMD mRNA Transcripts Analysis in a patient with Dystrophinopathy having a Nonsense Mutation. J Neurol Transl Neurosci 1: 1005.
INTRODUCTION
Duchenne muscular dystrophy (DMD, MIM# 310200) is caused by loss of the DMD-encoded sarcolemmal protein, dystrophin. Recently, an exon-skipping approach using antisense oligonucleotides, was shown to increase the expression of dystrophin and restore muscle function [1,2]. The approach targets recovery of the disrupted open reading frame and restoration of dystrophin expression, converting DMD to Becker muscular dystrophy (BMD, MIM#300376) or an asymptomatic phenotype. Nonsense mutations in DMD account for approximately 15% mutations in patients with dystrophinopathy and usually lack the dystrophin, resulting in the DMD phenotype. However, less than 5% of the BMD patients are known to carry nonsense mutations and possess a functional dystrophin protein [3-6]. This situation probably makes it difficult to predict phenotypes from the genotype in young patients. We report a new case of a patient with a nonsense mutation in DMD. Reverse transcription polymerase chain reaction (RT-PCR) analysis of DMD using biopsied skeletal muscle can help in assessing the phenotype.
PATIENT
The patient, now an 8-year-old boy, was the second child born to Japanese parents. His maternal great-grandfather exhibited gait difficulty of an undetermined origin in middle age. His maternal grandmother died of acute lymphoblastic leukemia without muscular symptoms. His mother showed no clinical manifestations. At the age of 1 year, the patient presented with asymptomatic hypercreatinine kinasemia at the time of admission for acute bronchitis, but he showed no muscle weakness or delayed developmental milestones. A skeletal muscle biopsy with hematoxylin–eosin staining revealed a few necrotic and regenerating fibers of various sizes. Immunohistochemical staining for dystrophin showed no signals in the sarcolemma. Based on these results, he was diagnosed as having DMD. He began walking at the age of 18 months. At the previous presentation to our clinic, he was 8 years old, and could walk to school and climb stairs. Neurological examination revealed that manual muscle testing scores were almost normal without calf hypertrophy. Blood examination revealed consistently elevated levels of serum creatine kinase, 3,380 IU/L (normal range: 62– 287 IU/L)” Cardiac echocardiography and electrocardiography showed normal findings.
METHODS
After obtaining informed consent, genomic DNA was extracted from white blood cells using the Gentra Puregene Blood Kit (Qiagen Inc. , Valencia, CA). Molecular analysis of intragenic deletions and duplications in the DMD was performed by the multiplex ligation-dependent probe amplification analysis (MLPA) for DMD using SALSA MLPA KIT P034/035 DMD /Becker (MRC-Holland, Amsterdam, Netherlands). Polymerase chain reaction (PCR) of exon 31 and the exon-intron boundaries of the DMD was performed with primers based on Leiden Muscular Dystrophy data pages (http://www. DMD. nl/). Total mRNA and total protein were extracted from the frozen skeletal muscle biopsied previously and synthesized cDNA using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). RT-PCR analysis of dystrophin mRNA from exon 27 to 32 used a primer set described previously [6]. Western blot analysis of dystrophin was performed by standard procedures using anti NCL-DYS2 (Leica Biosystems, Newcastle, UK). Immunohistochemistry of previously biopsied skeletal muscles was performed by standard procedures using anti-dystrophin antibody, NCL-DYS1, NCL-DYS2, and NCL-DYS3, and anti-utrophin antibody, NCL-DRP2 (Leica Biosystems, Newcastle, UK).
RESULTS
MLPA showed a normal exon dose, except for a slightly reduced probe signal in exon 31 (Figure 1).
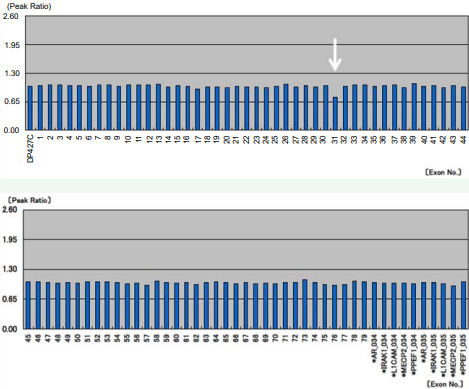
Figure 1 Multiplex ligation-dependent probe amplification analysis of DMD was performed using the patient’s DNA. The peak ratio of every exon was identified within a set range, but the signal of exon 31 was slightly reduced (white arrow).
The Patient’s genomic DNA was detected a truncating mutation in exon 31 of DMD (c. 4294C > T; p. Gln1432X) (Figure 2A).
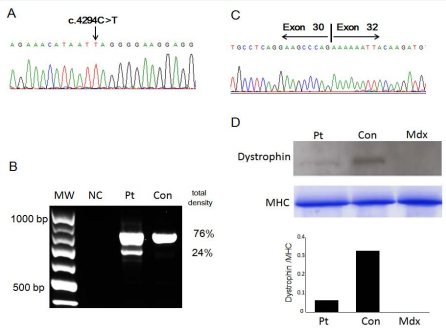
Figure 2 Genetic analysis of the patient. A: The DMD gene in genomic DNA was sequenced. Black arrow shows nonsense mutation in DMD. B: RT-PCR analysis of dystrophin mRNA from exons 27–32. RT-PCR products revealed two bands. The upper band was of expected size, 793 bp. The lower band was of a shorter size, 682 bp. The density of gel bands was analyzed using NIH Image (http://rsb.info.nih.gov/nih-image/). Density proportion was calculated on the basis of a total value of 100%. MW: molecular–weight marker (100–base ladder); NC: no template control; Pt: patient; Con: control. C: Sequence analysis of the shorter band revealed that patient’s cDNA lacked exon 31 in DMD. D: Upper: Western blot of total protein extract from skeletal muscle of control (Con), patient (Pt) and mouse model for muscular dystrophy (Mdx). 25 μg of protein were loaded in each lane. The blot was probed with NCL-DYS2 antibody. Middle: Control probe was using anti-myosin heavy chain (MHC). Lower: comparison of relative integrated density value of the bands carried out using an NIH Image revealed a 15% dystrophin content of normal control sample.
His mother was shown to be heterozygous for the mutation. The mutation was confirmed using mRNA derived from the previously biopsied skeletal muscles. RT-PCR analysis of dystrophin mRNA around exon 31 revealed an additional small-sized product present at 24% of the total density (Figure 2B). Direct sequence analysis of the additional product revealed a conjunction of exons 30 and 32, indicating the lack of exon 31 (Figure 2C). Western blot analysis of dystrophin revealed that the level of patient’s dystrophin was expressed as 15% of control (Figure 2D). Considering that dystrophin–lacking exon 31 is an in-frame product, we re-evaluated the immunohistochemistry and found that skeletal muscles were slightly stained with anti-dystrophin antibody and were occasionally accompanied by negative fibers (Figure 3A).
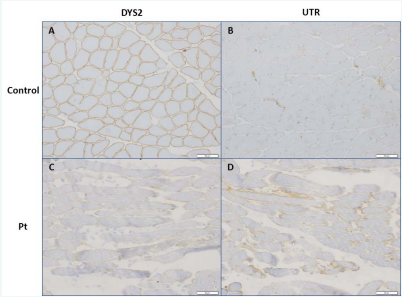
Figure 3 Immunohistochemistry of skeletal tissue in a control (A, B) and the patient (C, D). Skeletal muscles were stained with anti-dystrophin antibody (A, C) and anti-utrophin antibody (B, D).
In immunohistochemistry, utrophin-positive fibers were occasionally seen (Figure 3B). On the basis of these results, the patient was considered to present BMD phenotype.
DISCUSSION
We report a patient with BMD, possessing a nonsense mutation in the DMD gene. The dystrophin mRNA expression by using RT-PCR analysis showed 24% mRNA lacked of exon 31. The mRNA is expected to be partially functional dystrophin. Western blot analysis revealed total 15% expression of dystrophin in the patient. These results suggested that the measurements of dystrophin mRNA expression by using RT-PCR might reflect the quantification of dystrophin and substitute for western blot analysis. However, further study is needed to clarify whether the exon-skipping efficiency is different among muscles or not.
A patient with intermediate phenotype was reported of having partial in-frame skipping of exon 31 in a cell model, which was caused by the disruption of exonic splicing enhancer (ESE) or creation of exonic splicing silencer (ESS) motifs [3]. Another patient with a nonsense mutation in exon 31 was confirmed to have a limited amount of functional dystrophin in biopsied skeletal muscle cells [6]. And it was presumed that the mutations modified ESE/ESS motifs and promoted exon skipping. It was also reported that the small molecule TG003 induced skipping of exon 31 in a dose-dependent manner and increased the production of the dystrophin protein in cultured muscle cells [6]. Prediction of ESEs or ESSs in nonsense mutations for in-frame exons associated with BMD revealed that the c. 4294C > T mutation identified in our study was disrupted four ESE sites and created three ESS sites [7]. Thus, the mutation in our patient may modify exonic splicing regulatory site and promote skipping of exon 31.
CONCLUSIONS
This study reports a patient with BMD having a nonsense mutation in the DMDgene. The genetic and immunohistochemical results showed that the mutation led to the production of an in-frame transcript lacking exon 31. A subset of transcripts express as partially functional dystrophin. RT- PCR analysis in the DMD gene can be an easy-to-use and effective tool for prognosis of the clinical phenotype in a patient with a nonsense mutation. Recent advance in genetic analysis permit early diagnosis of dystrophinopathy in young patients. However, diagnostic errors are inevitable, considering the limited pathological or genetic testing. Testing using combined assays is required for definite diagnosis of patients with a DMD nonsense mutation.
ACKNOWLEDGMENTS
The authors appreciate the patients and their families for their cooperation. The authors are grateful to members of SHINSHU DMD WORKING GROUP for the support and SRL for providing image. This work was supported by Research on Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan #21300157 (AN), #24791844 (YN), and Shinshu Public Utility Foundation for Promotion of Medical Sciences (AN).









































































































































{kind=link}