Axonal Transport an Coats’ Disease and Congenital Oculodentaldigital Syndrome
- 1. Department of Ophthalmology, University of Southern California, Irvine, USA
Abstract
Purpose: We report axonal transport block in the lamina cribrosa (LC) of a two-year-old male with Coats’ disease in his right eye (Case 1), enucleated as blind and painful after an acute onset angle closure glaucoma. Two eyes from Case 2, a congenital Oculodentaldigital Syndrome (ODDS), are included as controls for the Coats’ case immunohistochemistry (IHC) and to report additional findings in this rare disorder.
Methods: We reviewed available history and performed histopathology on three globes including routine staining of paraffin-processed tissues and an IHC marker of orthograde axonal transport, APP-A4. Two eyes from a second 10-month-old infant with ODDS and normal IOPs served as controls for background staining with APP-A4.
Results: In addition to typical pathology for Coats’, the lamina cribrosa portion of the optic nerve from Case 1 demonstrated marked orthograde axonal transport block. Both eyes from Case 2 with ODDS had a history of congenital glaucoma treated medically before bilateral trabeculotomies at five months-age with normalization of IOPs. Descemet scrolls found OD are judged residual to goniotomy. Both globes from Case 2 demonstrated multiple anomalies previously reported in ODDS including micropthalmos, optic disc dysplasia OD, and persistent hyperplastic primary vitreous (PHPV) OU. Our case had also spontaneous retinal detachment OD prompting an exam under general anesthesia when, during induction, she expired with malignant tachycardia. Retinal ganglion cells (RCGs) labeled with APP-A4 OU, but optic nerve LC axons demonstrated only background accumulations of the IHC marker.
Conclusions: Orthograde axonal transport was interrupted in the LC of the Coats’ nerve due to inflammatory and neovascular angle closure reinforcing the importance of the LC as the site of initial optic nerve damage in glaucoma. The primary importance of the two ODDS globes for our report was as comparison controls for background APP-A4 staining in the Coats’ case. Our additional findings in the ODDS eyes suggest delayed spontaneous retinal detachment, and lens epithelial abnormalities are additional anomalies to add to the list of known malformations in this rare disorder. Case 2 also serves as a caution in ODDS regarding vulnerability to combinations of medications when general anesthesia is contemplated.
Citation
Minckler D, Stamper R, Han Y, Mosaed S, Lin K (2023) Axonal Transport an Coats’ Disease and Congenital Oculodentaldigital Syndrome. J Neurol Transl Neurosci 8(1): 1091.
CASE 1: COAT’S DISEASE
This two-year old male who had been otherwise healthy suffered an acute attack of pain OD with no light perception (NLP) and corneal edema preventing fundus exam. Clinical crisis records included a Tono-pen pressure of 20 mmHg OD, likely falsely low due to corneal edema artifact. Enucleation was performed promptly based on concern for possible retinoblastoma [1,2].
Pathology and IHC
The globe, received after several days in standard formalin solution, was confirmed as a right eye by the inferior oblique insertion. The cornea measured 10 x 10 mm horizontally and vertically and was grossly clear. The globe’s axial length was 20 mm, the vertical and horizontal dimensions were equal at 20 mm. There was no transillumination. The optic nerve stump measured 5 mm in length and 4 mm in diameter including the dural sheath. A 1 mm long-cross-section separated at the sclera along with the remaining longitudinal portions of the optic nerve was submitted separately. A standard horizontal PO section was accomplished without inducing obvious anterior segment distortion or lens displacement with a microtome blade cutting anteriorly from the superior edge of the optic nerve through sclera to the anterior vitreous cavity paralleling the cutting board surface before rotating the globe and blade perpendicular to the surface to complete the cut. The inferior and superior globe caps were cut free and left in fixative. The optic nerve samples and the PO block with disc side down were submitted for routine paraffin processing and staining with H&E, PAS, Iron, trichrome, GFAP, CD68, CD68 red, and APP-A4. Standard instructions to histology technicians regarding the PO block are to aim for stained sections to include the central portions of the disc.
Microscopic Description
H&E-stained sections include the optic disc and scleral optic nerve. The cornea is histologically unremarkable with good preservation of endothelium. The anterior chamber angles are partially closed by variable peripheral anterior synechias for an estimated 10-20 percent of both sides of the meshwork most advanced temporally. CD68 positive (CD68+) and CD68 red+ macrophages infiltrate the meshwork (Case 1 Figure 1 B1, B2, Figure 3 A) and line the iris surface. Ectropion uveae is present on the pupil edges most pronounced temporally. Iris neovascularization is apparent most obviously along the temporal pupil edge (Figure 1 B2). Massive numbers of CD68+ and CD68 red+ macrophages line both leaves of the anterior iris surface, infiltrate the trabecular meshwork and are accumulated within zonular fibers at both lens equatorial edges (Case 1 Figure 1 A1).
![An Enlarged scan of H&E-stained PO section revealing retinal detachment and eosinophilic subretinal exudate filling the subretinal space. The lens is distorted posteriorly suggesting pressure from behind (H&E original magnification X 1); A 1 Photomicrographs of macrophages in anterior vitreous at the lens edges (H&E original magnification X 200); A 2 normal appearing LC [blue bracket] (H&E original magnification 32.25); B Iris and temporal angle with CD68+ macrophages lining iris surface and infiltrating meshwork with approximately 20% angle closure by PAS in this view. Enlarged Schwalbe’s line [blue arrow] marks the end of Descemet’s (H&E original magnification X 125); B 2 Temporal iris edge with ectropion uvea and NV membrane [blue arrow] (original magnification X 200); B 3 Optic nerve at different plane than A 2 with APP-A4 reaction product prominent in LC axon bundles (original magnification X 31.25); C APP-A4 positive RGCs from relatively well preserved retinal fragment (APP-A4 original magnification X 500); C 1 High magnification photomicrograph of LC from portion of B 3 demonstrating end bulb swellings along axon profiles (APP-A4 original magnification X 787.5 oil immersion).](https://www.jscimedcentral.com/public/assets/images/uploads/image-1692683002-1.PNG )
Figure 1: An Enlarged scan of H&E-stained PO section revealing retinal detachment and eosinophilic subretinal exudate filling the subretinal space. The lens is distorted posteriorly suggesting pressure from behind (H&E original magnification X 1); A 1 Photomicrographs of macrophages in anterior vitreous at the lens edges (H&E original magnification X 200); A 2 normal appearing LC [blue bracket] (H&E original magnification 32.25); B Iris and temporal angle with CD68+ macrophages lining iris surface and infiltrating meshwork with approximately 20% angle closure by PAS in this view. Enlarged Schwalbe’s line [blue arrow] marks the end of Descemet’s (H&E original magnification X 125); B 2 Temporal iris edge with ectropion uvea and NV membrane [blue arrow] (original magnification X 200); B 3 Optic nerve at different plane than A 2 with APP-A4 reaction product prominent in LC axon bundles (original magnification X 31.25); C APP-A4 positive RGCs from relatively well preserved retinal fragment (APP-A4 original magnification X 500); C 1 High magnification photomicrograph of LC from portion of B 3 demonstrating end bulb swellings along axon profiles (APP-A4 original magnification X 787.5 oil immersion).
Hemorrhage is absent in the eye except for small foci of degenerated blood in the subretinal exudate and no inflammatory cells other than macrophages are recognized. Iron stains are negative. The lens is in normal position and the capsule intact (Case 1 Figure A1). Lens epithelial cells appear normally distributed nasally but have migrated more posteriorly than normal temporally. The posterior nasal lens is abnormally swollen, or perhaps indented, as the misshape suggests posterior pressure. The vitreous posterior to the lens equator is clear. A complete funnel shaped, partially loculated retinal detachment is present emanating from the optic disc. Retinal architecture is degenerated especially in the periphery where normal cell layers cannot be distinguished (Case 1 Figure 2 A-B, Figure 3 B-C).
![A Degenerated retina with no photoreceptor or outer segment layers demonstrating myriad invaded macrophages, some with visible nuclei, within residual MÜeller cell fibers extending from their fusion with the ILM to the external limiting membrane (ELM) fibroblasts are layered in spaces between MÜeller fibers in the position of former RGCs and the inner plexiform spaces (APP-A4 original magnification X125); B Low power view of degenerated retina between subretinal exudate collections. The nerve fiber layer (NFL) (blue bracket) is expanded by layers of fibroblasts anterior to faint profiles of persisting RGCs, and the middle and outer nuclear layers. Subretinal fibrosis, presumed from fibrous metaplasia of RPE, lines the outer retina profile (H&E original magnification X 31.25, 50 µm scale bar) [inset: cholesterol crystal spaces within exudate (H&E original magnification X 500]; C Dalen-Fuch’s-like nodule of altered RPE on Bruch’s membrane near the disc (H&E original magnification 31.25); [inset: H&E stain of same nodule at higher original magnification X500]; D High magnification of partly depigmented RPE, choriocapillaris with RBC (blue arrow) and inner choroid. Bruch’s membrane is not discernable (H&E original magnification X 787.5 oil immersion, 10 µm scale bar); E Highly magnified pigment clumps from exudate near RPE, (H&E original magnification X 787.5 oil immersion, 2 µm scale bar).](https://www.jscimedcentral.com/public/assets/images/uploads/image-1692683038-1.PNG)
Figure 2: A Degenerated retina with no photoreceptor or outer segment layers demonstrating myriad invaded macrophages, some with visible nuclei, within residual MÜeller cell fibers extending from their fusion with the ILM to the external limiting membrane (ELM) fibroblasts are layered in spaces between MÜeller fibers in the position of former RGCs and the inner plexiform spaces (APP-A4 original magnification X125); B Low power view of degenerated retina between subretinal exudate collections. The nerve fiber layer (NFL) (blue bracket) is expanded by layers of fibroblasts anterior to faint profiles of persisting RGCs, and the middle and outer nuclear layers. Subretinal fibrosis, presumed from fibrous metaplasia of RPE, lines the outer retina profile (H&E original magnification X 31.25, 50 µm scale bar) [inset: cholesterol crystal spaces within exudate (H&E original magnification X 500]; C Dalen-Fuch’s-like nodule of altered RPE on Bruch’s membrane near the disc (H&E original magnification 31.25); [inset: H&E stain of same nodule at higher original magnification X500]; D High magnification of partly depigmented RPE, choriocapillaris with RBC (blue arrow) and inner choroid. Bruch’s membrane is not discernable (H&E original magnification X 787.5 oil immersion, 10 µm scale bar); E Highly magnified pigment clumps from exudate near RPE, (H&E original magnification X 787.5 oil immersion, 2 µm scale bar).
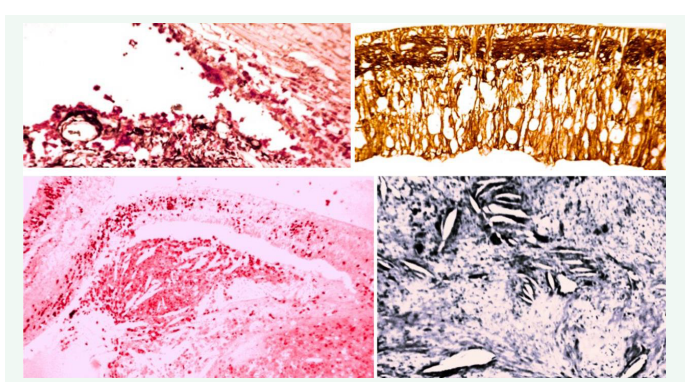
Figure 3: A CD68 red+ macrophages infiltrating the angle and lining the iris surface (original magnification x 125); B Degenerating retina with loss of all layers of RGCs and marked lamellar gliosis intertwined between MÜeller cell remnants extending from ILM to ELM (Glial Acid Fibrillary Protein stain original magnification x 125); C CD68 red+ macrophages infiltrating retinal remnants and cholesterol spaces in subretinal exudate (original magnification x 31.25); D Mason Trichrome stain better demonstrating cholesterol crystal spaces in subretinal exudate (original magnification x 125).
Posteriorly, near the disc cell layers retain some normal populations but outer segments are absent. Only a few retinal vessels of small size are found containing blood. Degenerating retina near the disc, especially in APP-A4 IHC stains, have prominent Mueller cell processes blending with the internal limiting membrane (Case 1 Figure 2A, Figure 3B). Macrophages are prominent within all discernable retinal layers (Figure 2A). Huge numbers of vacuolated partly necrotic degenerating cells, presumably macrophages, accumulate along remnants of the retina. A massive eosinophilic exudate fills most of the subretinal space and contains huge clusters of empty cholesterol crystals (Case 1 Figure 2B inset), many surrounded by CD68+ macrophages, some including cytoplasmic pigment. Focal accumulations of degenerated blood and clusters of cells containing round to oblong pigment granules of 2-3-micron diameter, possibly of retinal pigment cell (RPE) origin, are also present in the exudate (Case 1 Figure 2D, E). Large segments of the RPE demonstrate degeneration or cell loss. Bruch’s membrane and red cell-containing choriocapillaris vessels were present only in a few areas (Case 1 Figure 2D). Variable similar pigment granules in macrophage clusters like those in the subretinal exudate are scattered throughout the choroid most prominently near vessels.
APP-A4 Immunohistochemistry (IHC)
Axonal transport is essential for normal growth and function of the mammalian nervous system [4-10]. The reaction product accumulation is prominent in lamina cribrosa axonal bundles in our Coats’ case, consistent with block of orthograde transport identical to results in experimental studies of ocular hypertension in foveated primates (Figure 1B 3, C1) [4-10]. And in abusive head trauma (AHT) [11]. Routine H&E and PAS stains of the LC in case 1 appear normal without a hint of pathology [6] (Case 1 Figure 1 A2).
CASE 2 ODDS
A 5-month-old baby girl who was born at 36 weeks gestation was noted to have cloudy corneas, tearing and extreme sensitivity to light since birth in both eyes. She was diagnosed with congenital glaucoma and eventually with oculodentaldigital dysplasia. She had syndactyly of the 4th and 5th fingers of both hands. The mother denied any pregnancy difficulties other than low amniotic fluid necessitating induction of labor and had no history of prior birth abnormalities, infections, or substance abuse. The baby’s first glaucoma therapy was latanoprost once a day and dorzolamide-timolol combination twice daily OU. Examination under anesthesia (EUA) in July 2011 revealed elevated intraocular pressure (IOP) via pneumotonometer of 36 mmHg in her right eye and 27mmHg in her left eye with. Her corneas were edematous. Her horizontal corneal diameter (CD) was 9.4 mm in both eyes and the central corneal thicknesses were 674 microns in the right eye (OD) and 677 microns in the left eye (OS) with central Haab’s stria in both eyes (OU). Persisting vitreous vessels at the posterior capsule of the lens were noted OU with vertical cup-to-disc ratios (CD) of 0.8 in both eyes. Gonioscopic exam revealed open angles with moderate meshwork pigmentation OU. The retinas were normal OU. The working diagnosis was primary congenital glaucoma. She then underwent ab externo trabeculotomy OD with the ISCIENCE iTrack 250A catheter. At approximately 180 degrees of cannulation of Schlemm canal, the catheter encountered an obstruction and could not be passed further. Passage in the opposite direction showed a blockage at approximately the same location. Therefore, only 180 degrees of trabeculotomy were accomplished. One week later, the left eye underwent an ab externo attempt at 360-degree trabeculotomy with the same type of catheter but at approximately 25% of the canal circumference the cannula tip deviated posteriorly. Efforts to cannulate in the opposite direction were met with similar results. Therefore, a Harms trabeculotomy probe was used to perform a trabeculotomy ab-externo in each direction from the incision site over approximately 40% of the superior trabecular meshwork. Her last EUA was performed 4 months after the second surgery when IOPs were 15 mmHg OD and 23 mm Hg OS. Corneal diameters were measured at 8.5 mm OU, smaller than previously of unknown significance since CD is a variable measure. Her corneas were still slightly cloudy OU but and her cup-to-disc ratios were decreased to 0.2 in both eyes. Dilated fundus exam demonstrated the retina to be flat and unremarkable bilaterally. She was to continue no anti-glaucoma drops OD and dorzolamide-timolol OS but was lost to follow-up thereafter.
At ten months age, a spontaneous right retinal detachment was recognized and thought by a retina specialist as possibly related to persistent hyperplastic primary vitreous (PHPV) [3] She was scheduled for exam under general anesthesia to include fundus photography, fluorescein angiography, a pars plana vitrectomy with retinal laser, lensectomy, membrane peel, detachment repair and oil injection. During attempted anesthesia she developed uncontrollable malignant trachycardia and expired despite attempted resusscitation with no surgery performed. A partial clinical exam had revealed remnants of the tunica vasculosa lentis and elongated ciliary processes consistent with PHPV. Autopsy was performed by the Los Angeles Coroner and her eyes referred to UCI for ocular pathology examination.
Both globes were received, accurately labeled in separate bottles (A right and B left) after 14 days in formalin. The corneas were and 9 mm in horizontal diameter and clear bilaterally. Transillumination was normal OU. Axial lengths were 16 mm OD and 20 OS with 15 mm horizontally and vertically OD and 19 mm vertically and 18 mm horizontally OS. The pupils were approximately 3 mm in diameter and slightly irregular OU. The optic nerve stumps measured 21 mm in length and 3 mm in diameter OD and 17 mm in length and 3 mm in diameter OS. The right eye was opened in a standard PO fashion revealing a total funnel retinal detachment without grossly obvious retinal hemorrhages or cysts (case 2 right eye A1). A PO section of the left globe revealed an attached retina and clear vitreous, free of hemorrhage. The left eye sustained substantial mechanical damage during the gross PO preparation (case 2 left eye A1). PO sections of both globes and cross and longitudinal sections of optic nerves were submitted for paraffin processing and staining to include, H&E, PAS, GFAP and APP-A4.
The PO sections OD include central sectioning of the optic nerve head demonstrating microscopic vacuolization suggesting fixation artifact (Figure 4 case 2 A1).
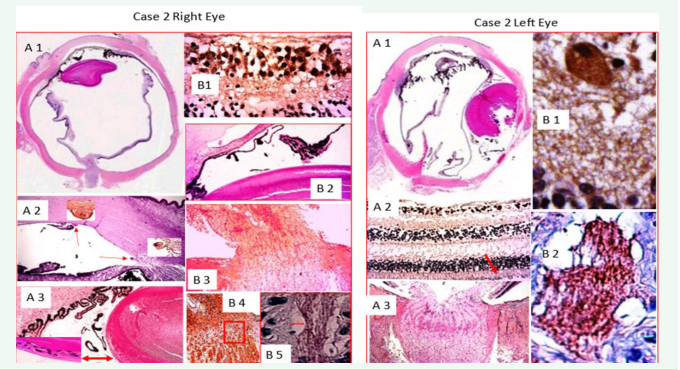
Figure 4: A 1 is an enlarged scan of the PO section reveals abnormal pigmentation extending along the endothelium, a distorted lens, peripheral retinal contracted folds near pars plana and optic disk dysplasia (H&E original magnification X 1); A2 displays the anterior segment and open angle with two Descemet’s scrolls one central and one in the angle residual to prior trabeculotomy; insets at higher magnification provide better details. (H&E original magnifications X 31.25 and 125); A 3 Low power image of nasal anterior chamber, ciliary body with elongated processes, and an intact lens capsule but anomalous multi-layered lens epithelium posterior to the equator, the inset provides a higher magnification of the abnormal lens epithelium (H&E original magnifications 31.25 and 125); B 1 APP-A4 labeled macular RGCs (APP-A4 original magnification X125); B 2 Nasal angle with partial PAS and abnormal elongated ciliary process extending along posterior iris (H&E original magnification X 31.25); B 3 Dysplastic optic disc and retinal detachment stalk (H&E original magnification 31.25); B 4 Low power photo of lamina cribrosa with axonal columns interspersed with glial-collagen beams (APP-A4 original magnification X 31.25); B 5 High power magnification of area in B 4 red box illustrating background APP-A4 background labeling of LC axonal bundles (APP-A4 original magnification X 787.5 oil immersion with 5 µm scale bar)
The cornea OD is remarkable for focal loss of endothelium and Descemet’s scrolls, one centrally and one in the angle (Figure 4 case 2 right eye A2 insets). PAS are present nasally, probably residual to goniotomy. The sclera is intact. The lens has an intact capsule OD but is distorted with a bulge posteriorly of uncertain cause. There is posterior migration of lens epithelium, some in two layers, posterior to the equator especially nasally associated with elongated ciliary processes (Figure 4 case 2 right eye A2, A3). Minimal remnants of persisting primary vitreous vessels are present near the lens surfaces (Figure 4 case 2A 3 B2). Autolysis of the retina near the disc temporally is present with loss of photoreceptor outer segments, consistent with delay in fixation. A delicate preretinal membrane is present far peripherally nasally along the retinal surface. Marked retinal folding with adhesions between the internal limiting membranes (ILMs) of adjacent folds both nasally and temporally likely explain a spontaneous circumferential contraction and retinal detachment. Mononuclear cells, probably macrophages, are trapped between some folds. The RPE remains attached to the posterior pars plana just anterior to the retinal folds temporally and nasally. No fibrovascular tissues are found connecting the retinal folds to the ciliary body or posterior lens capsule. Choroidal vessels are markedly congested. Retinal ganglion cells are diminished especially temporally. The LC OD is still recognizable in its normal position adjacent to the sclera even though the disc is tilted and dysplastic. APP-A4 label is definite in RGCs in detached macular retina but only background label is present in the LC.
The cornea OS is unremarkable with good preservation of endothelium. Partial angle closure by PAS may be present nasally but artifact complicates assessment. The iris is fragmented, but free of neovascularization, and elongated pigmented ciliary processes line its posterior surface. The lens OS was mechanically dislocated nasally with partial capsule and cortex loss. Minimal fibrous remnants of the tunica vasculosa lentis remain along the lens capsule similar-to those OD. Some fragments of retina with relatively well-preserved cell details demonstrate RPE pigment retention indicating some areas of persisting retinal attachment at fixation (Figure 5 case 2 left eye A2). APP-A4 positive RGCs are present in retinal fragments but axons in the LC demonstrate only background staining visible at high magnification (Figure 5 case 2 B1, B2).
![A 1 Enlarged scan of the left eye PO section with prominent fragments of abnormally elongated pigmented ciliary processes scattered across the interciliary space. The lens is dislocated nasally and fragmented posteriorly; A 2 fragment of macular retina with APP-A4 positive RGCs and outer segments retaining portions of RPE pigment [red arrow] (H&E original magnification X 125); A 3 normal appearing optic nerve head with LC visible as curving line of fibro-glial boundaries between axonal bundles (H&E original magnification X 31.25); B 1 large APP-A4 positive RGC (APP-A4 original magnification X 500); B 2 High magnification of background axonal bundle labeling in lamina cribrosa (APP-A4 original magnification X 787.5 oil immersion).](https://www.jscimedcentral.com/public/assets/images/uploads/image-1692683100-1.PNG)
Figure 5: A 1 Enlarged scan of the left eye PO section with prominent fragments of abnormally elongated pigmented ciliary processes scattered across the interciliary space. The lens is dislocated nasally and fragmented posteriorly; A 2 fragment of macular retina with APP-A4 positive RGCs and outer segments retaining portions of RPE pigment [red arrow] (H&E original magnification X 125); A 3 normal appearing optic nerve head with LC visible as curving line of fibro-glial boundaries between axonal bundles (H&E original magnification X 31.25); B 1 large APP-A4 positive RGC (APP-A4 original magnification X 500); B 2 High magnification of background axonal bundle labeling in lamina cribrosa (APP-A4 original magnification X 787.5 oil immersion).
CONCLUSIONS
Our findings in the Coat’s case include typical histopathology long described in this disorder including cholesterol crystals in a massive exudative retinal detachment with anterior chamber migration of macrophages carrying debris from retinal degeneration to the outflow pathway with iris neovascularization and partial angle closure, (Figure 1, Case 1) [2]. NFL expansion and subretinal fibrosis are presumed residual to sclerosis of telangiectactic vessels and metaplasia of RPE. Retinal degeneration was advanced perhaps explaining our not finding abnormal retinal vessels. Dense staining of Mueller cells by APP-A4 (Figure 2A Case 1) was not anticipated and may be useful in other ongoing histology studies. The important new finding is clear evidence of LC block of orthograde axonal transport in congenital glaucoma reinforcing conclusions from experimental and previously published studies [4-11,12]. While Case 2 ODDS specimens were of interest primarily as background staining controls for lamina cribrosa block of axonal transport, we can suggest that spontaneous retinal detachment and layered posteriorly migrated lens epithelium OD may be additional anomalies encountered in this rare disorder[3, 13].1 Angle closure in one or both eyes could have been residual to goniotomy.
REFERENCES
- Spencer WH. Ophthalmic Pathology: An Atlas and Textbook, Volume-2 Third Ed. American Academy of Ophthalmology, Saunders, New York. 1985.
- Coats’ Disease. 625-634
- Persistent Hyperplastic Primary Vitreous (PHPV). 559.
- Bunt AH and Minckler DS, Jakobiec FA. Ocular Anatomy Embryology and Teratology. CH 21 Axonal Transport Basic Aspects. 1982; 639- 549.
- Minckler DS, Bunt AH, Jakobiec FA. Ocular Anatomy Embryology and Teratology, CH 22 Axonal Transport clinical Aspects. 1982; 651-675.
- Schliwa M, Woehlke G, Molecular motors. Insight Review, Nature. 2003; 422: 759-765.
- Minckler DS, Tso MO, Zimmerman LE. A light microscopic, autoradiographic study of axoplasmic transport in the optic nerve head during ocular hypotony, increased intraocular pressure, and papilledema. Am J Ophthalmol. 1976; 82:741-757.
- Minckler DS, Bunt AH, Klock IB. Radioautographic and cytochemical ultrastructural studies of axoplasmic transport in the monkey optic nerve head. Invest Ophthalmol Vis Sci. 1978; 17:33-50.
- Quigley H, Anderson DL. The dynamics and location of axonal transport blockage by acute intraocular pressure elevation in primate optic nerve. Invest Ophthalmol. 1976, 15; 606-616.
- Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest. Ophthalmol Vis Sci. 1977; 16: 426-441.
- Minckler DS, Charlson ES, Nalbandian A. Unique Pre-ß Amyloid Precursor Protein (APP-A4) Observations in Ocular Tissues from Suspected Abusive Head Trauma Victims. J Psychol Clin Psychiatry. 2017; 7: 00435.
- Minckler DS, Brown DJ, Nalbandian A, Suh DW. Amyloid Precursor Protein in Abusive Head Trauma Suspects, in-press Am J Ophthalmol. 2022; 240: 58-66.
- Kumar V, Couser NL, Pandya A. Oculodentodigital Dysplasia: A Case Report and Major Review of the Eye and Ocular Adnexa Features of 295 Reported Cases. Case Rep Ophthalmol Med. 2020; 2020: 6535974.







































































































































{kind=link}