Bulbar Predominant Amyotrophic Lateral Sclerosis with Bilateral Amygdala Atrophy and Vascular Leukoencepholpathy Showing Anxiety, Depression and Globally Preserved Cognition
- 1. Department of Geriatric Psychiatry, Klinikum Christophsbad, Germany
- 2. Department of Psychiatry and Psychotherapy, Klinikum Christophsbad, Germany
- 3. Department of Radiology and Neuroradiology, Klinikum Christophsbad, Germany
Abstract
We here report a peculiar case of a 76 year old woman with the bulbar form of amyotrophic lateral sclerosis presenting as a complex neuropsychiatric disorder with bilateral amygdala atrophy and a vascular leukoencepholpathy associated with anxiety/emotional disturbances and relatively intact cognition. In fact, her motor disorder was dominated by lower motor neuron signs in bulbar nuclei with pronounced dysarthria , medium severe dysphagia, elevated anxiety values, and depressed mood. Global cognitive function was unremarkable. Magnetic resonance imaging revealed significant atrophy of the amygdala and adjacent parts of the mediotemporal lobe cortex bilaterally (right greater than left), a slightly rounded/enlarged ventricular system (esp. the tips of temporal horns), a widening of the subarachnoid space temporo-polar, and a widespread leucoencephalopathy.
Keywords
• Amyotrophic lateral sclerosis
• Amygdala
• Atrophy
• Leukoencephalopathy
Citation
Geser F, Hermle L, Egan PJ, Mitrovics TCG (2015) Bulbar Predominant Amyotrophic Lateral Sclerosis with Bilateral Amygdala Atrophy and Vascular Leukoencepholpathy Showing Anxiety, Depression and Globally Preserved Cognition. J Neurol Transl Neurosci 3(1): 1054.
ABBREVIATIONS
ALS: Amyotrophic Lateral Sclerosis; MRI: Magnetic Resonance Imaging
INTRODUCTION
The potential use of a ‘‘bio-marker’’ is based on the idea that pathophysiologic processes, which lead to structural and functional abnormalities, are accompanied by a variety of biological changes. A validated and sensitive biomarker would offer significant ante-mortem diagnostic and clinical benefits - it would provide the potential to assess the effect of therapeutic interventions. Since there is no firm diagnostic ante-mortem tool for ALS so far [1], an in vivo biomarker-focused approach is now - as stated by the first Neuroimaging Symposium in ALS - a research priority [2].
CASE PRESENTATION
Here we report on a 76 year old woman with a prior history of hypertension, hyperlipidemia, bilateral gonarthrosis (with a cartilage transplantation done in 2002 on the right side and a knee prosthesis implanted in 2013 on the left side), hypothyreosis, and coronary artery disease (with 2 stents implanted in 2009). Furthermore, the patient had a cornea transplantation done in 1999 (left side). She reported to be severely hearing impaired on the left side since childhood. Starting in late 2013, the patient developed progressive difficulties with speaking and swallowing as well as depressed mood. She also developed a medium-severe episode of depression which was treated with escitalopram. At the time we first saw her at our hospital, a diagnosis of probable ALS was made. Her motor disorder was dominated by lower motor neuron signs in bulbar nuclei with pronounced dysarthria and medium-severe dysphagia. In fact, a video fluoroscopic examination showed evidence of a dysphagia with weak tongue movements, and weak, slowed, and discoordinated swallowing. She reported to have more problems with liquid than solid food. Speech was severely dysarthric with an unclear, slurred quality. Her voice was weak (hypophonia) and barely understandable, so that she started to write things down on paper in order to communicate. No unequivocal other symptoms or signs of cranial nerve involvement (such as ocular motility) were seen. Electromyography testing revealed neurogenic changes in bulbar and cervical segments, and fasciculations, respectively. She showed signs of upper motor neuron dysfunction including exaggerated reflexes and broadening of reflex zones in the cervical and lumbar segments, respectively. Slight bilateral spasticity was found with the upper limbs. There was no Babinski sign and no clonus of the feet. A significant decrease in amplitude in somatosensory evoked potentials studies was seen in cervical segments. Motor evoked potential response latencies and the central motor conduction times to the lower, but not upper, limb were delayed. The patient did not report difficulties with sensation. Pallesthesia was tested by using a vibrating tuning fork placed forkplaced on the styloid process (7-8/8 bilaterally) and lateral malleolus (7/8 and 6/8 on the left and right side, respectively). Auditory evoked potentials showed evidence of a lesion of the retro-cochlear auditory pathway on the left side, and visual evoked potentials and the orbicularis oculi reflex were normal. There was no tremor or involuntary movements. She complained about an overall weakness, and there was instability with (tests of) stance or walking. She showed pathological crying (and laughing) and psychomotor slowness. She did not complain about difficulties with memory. Furthermore, no chances in here social activities or behavior were reported. However, her husband noted that she was listening to music or watching the television using head phones for “extended period of times”. The patient did not complain about bladder difficulty or permanent bowel difficulties. However, since about the last 10 years, she experienced fatigue, and for about the same time, she complained about slight problems with maintaining, but not initiating, sleep. Her family history was negative for neurological or psychiatric diseases. Analysis of the cerebrospinal fluid did not reveal any significant results. Structural magnetic resonance imaging (MRI) studiesshowed significant atrophy of the amygdala and adjacent parts of the mediotemporal lobe cortex (such as the periamygdaloid cortex) bilaterally (right greater than left, Figure and Supplemental Figure 1),
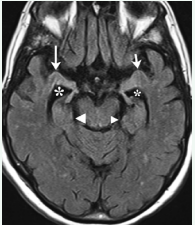
Figure 1 1.5 Tesla MRI, fluid attenuated inversion recovery sequence, axial plane, showing atrophic amygdala (right: large arrow, left: short arrow). For the smaller right and larger left hippocampus see large and small arrow head, respectively. The temporal horn of the right lateral ventricle (large asterisk) is slightly larger than the left one (small asterisk).
a rounding and enlargement, resp., of the supratentorial ventricular system (especially the tips of temporal horns, Figure, Supplemental Figures 1, 2, 3a, 3b, and 4a) and a widening of the subarachnoid space temporo-polar with atrophy (especially) of the tip of the temporal lobe (Supplemental Figures 4a and 4b). The right hippocampus and adjacent parts of the temporal lobe appeared to besomewhat smaller than the left ones Figure, Supplemental Figures 3a and 3b). There were widespread gliotic lesions in the deep white matter of the cerebral hemispheres with patchy subcortical (vascular leukoencepholpathy) and subependymal lesions (Supplemental Figure 2). In addition, a small lacunar defect was detected in the upper part of the right hemisphere of the cerebellum. Neuropsychological testing at our hospital was done at a time when she was treated with two anti-depressants (i.e., venlafaxine and mirtazapin). It showed elevated anxiety values as measured by the State-Trait Anxiety Inventory (trait-anxiety score 59, corresponding to the 95 percentile; state anxiety score: 67), mild depression according to the Beck Depression Inventory (score:14), and – apart from impairment in the learning of word lists – average scores for verbal and visual cognitive functions. The Consortium to Establish a Registry for Alzheimer’s Disease neuropsychological battery showed averages scores for verbal and visual cognitive functions - except for word learning, which was below average; the Mini-Mental State Examination scored 30. Global cognitive function was unremarkable (score in the clock drawing test: 2), but concentration was far below average. The patient complained transiently, i.e., at the time of initial diagnosis, about pain of the right side of the face and tongue and pain when swallowing. She also said to be tired of life, but there was no actual suicidality. The patient was put on riluzole and other attempts in her neuropsychiatric medication – in addition to her antidepressant medication – included quietapine, pregabalin, zolpidem and melperon. Within a short period of time, which was by mid of 2014, she became too weak to stand without assistance, and the disease progression of ALS necessitated the placing of a percutaneous endoscopic gastrostomy for nutrition. She got completely anarthric, and there was a further deterioration in her mood (crying) and writing (not legible).
DISCUSSION
Motor neuron disease, cognitive impairment as well as speech impairment have been known to co-occur since many years, and (clinico-) pathological studies corroborate the multisystem nature including the limbic system with overlapping features of this conditions [1]. Atrophy as measured by in vivo neuroimaging techniques mirror the neurodegenerative process and can thus be used to determine topographical pattern of degeneration and monitor rates of disease progression. Structural neuroimaging studies in ALS - for review see [3-6] and references therein - have yielded in varying, and in part, conflicting results such as atrophy of the spinal cord, motor/premotor cortex in some studies, and the grey matter in frontal, temporal, occipital lobes and limbic regions. Studies on ALS patients have shown that emotional disturbances (depression, anxiety, and “pseudobulbar” affect) do occur to a varying degree in this disorder [7-11]. The amygdala is known to play a pivotal role in emotion. Indeed, functional MRI has been used to show an activation of the amygdala/ periamygdaloid cortex during conditioned fear acquisition and extinction in normal subjects [12]. In patients with ALS, emotional responding has been shown to be altered towards a positive valence [13] suggesting involvement of the amygdala. A MRI volumetric study found a statistical trend towards lower total amygdala volume in the ALS as compared to control subjects [14]. We here show significant amygdala atrophy bilaterally (right greater than left) in a patient with bulbar predominant ALS and emotional disturbances. There also was a loss of volume of the mesiotemporal lobe as a whole and a widespread leukoencepholpathy. Widening of the subarachnoid space was especially visible at the tips of the temporal lobes. Cognitive dysfunction was not absolutely absent, but much less pronounced as compared to the motor and mood disorder. The latter two started at around the same time. Correspondingly, the morphological signs of atrophy of the cerebrum such as the rounding of the supratentorial ventricular system (esp. the tips of the temporal horns) and a widening of the subarachnoid space termporo-polar were there, but only to a slight degree. On functional imaging, ALS patients have been shown to display analtered left amygdala-prefrontal cortex connectivity [15]. In patients with the “temporal variant” of front temporal dementia, emotional comprehension has been show to correlate with atrophy in the right amygdala and the right orbitofrontal cortex [16]. For depression and anxiety, it is known that activation of the amygdala occurs and there may be both an increase and a decrease in amygdala volumes in these disorders. It has been, in fact, suggested that there is a time course in these volumes with larger amygdala occurring with the first episodes of depression (probably reflecting hyperactivation with higher metabolism and blood flow) than with subsequent, recurrent episodes [17]. As the diseases progresses, excitotoxic processes may result in a neurodegenerative process and finally in atrophy. In this respect, it is of interest that the anti-glutamate agent riluzole has at least a modest effect on survival in ALS. Furthermore, intoxication with homoic acid, which is structurally related to glutamate and has excitotoxic properties, has been followed by neuronal loss and gliosis predominantly in the hippocampus and amygdala after ingestion of contaminated mussels [18].
In conclusion, we here show marked atrophy of the amygdala with a pronounced right-left difference, a widespread vascular leukoencepholpathy, but no marked global neocortical atrophy, in a case with ALS clinically presenting mainly with dysarthria, dysphagia, anxiety, depression, and no major memory or behavioral changes. These findings indicate that the bulbar form of ALS can present as a complex neuropsychiatric disorder with bilateral amygdala atrophy and a vascular leukoencepholpathy neuroimaging associated with anxiety/emotional disturbances and relatively intact cognition.









































































































































{kind=link}