Obtaining Informed Consent via Videoconference: A Pilot Randomized Trial
- 1. Department of Emergency Medicine, University of Cincinnati, USA
Abstract
Background: In acute care research and research in remote environments, practical difficulties of having a researcher available to obtain informed consent can be a barrier to recruitment. Attempts have been made to overcome this using videoconferencing, but how this approach impacts the consent process is unknown.
Methods: sham trial comparing two FDA-approved medications for hypertension. Patients were randomized to be enrolled: 1) at the bedside or 2) via videoconferencing using Face Time on an iPad 2. Following the enrollment attempt, subjects were debriefed about the sham trial and the true study goals, and given Between March 2013 and October 2013, Emergency Department (ED) patients with elevated blood pressure were offered the opportunity to participate in a educational and referral information for hypertension.
Results: Twenty-one subjects were approached for enrollment in the sham trial, 10 at the bedside and 11 by videoconferencing. Mean age was 43 years (SD 11), 76% were African-American, 62% were male, and 81% had at least a high-school education. Consent rate for the sham trial was 91% by videoconferencing and 80% in-person (difference 11%, 95%CI -19% to 41%). No subject reported difficulties communicating with the physician, and there were no differences in understanding the components of the informed consent.
Conclusion: Consent rates, subject comfort level, and knowledge transfer during sham trial enrollment were similar when the process was done by videoconferencing versus in person. While limited to a small sample in an urban ED, findings suggest that videoconferencing may be used to facilitate physician-participant communication during informed consent for clinical trials.
Citation
Thomas R, McMullan J, Hart K, Lindsell CJ, Linke M, et al. (2015) Obtaining Informed Consent via Videoconference: A Pilot Randomized Trial. J Neurol Transl Neurosci 3(1): 1056
Keywords
• Clinical research
• Videoconference
• Telemedicine
• Informed consent
• Research ethics
INTRODUCTION
A core element of the process of informed consent in clinical trials is to provide the participant information about the study procedures followed by an opportunity to ask questions about the study, the treatments, and the alternatives. Research participants often do not understand the information they are provided during the informed consent procedure [1]. In acute care research involving medical or procedural interventions in acutely ill patients, physicians are often involved in obtaining informed consent.
Audio-visual telemedicine or videoconferencing has become well-established in clinical care for overcoming the barriers of bringing the expert clinician to the bedside and it is commonly used in acute stroke, trauma, prehospital care and non-English medical translation services [2-4]. Primary care clinics have also had successful results with the use of videoconferencing in conducting interviews with care management. Patients indicated a high degree of satisfaction and expressed willingness to recommend videoconferencing to others [5].
Currently, there are no regulations available regarding the use of videoconference for obtaining informed consent in clinical research in an acute care setting. Nonetheless, investigators have begun using videoconferencing for enrolling patients in clinical trials [2,6-8]. The primary aim of this pilot investigation was to determine if any differences in rate of consent existed using the videoconferencing approach versus the physician at bedside approach. Secondary aims were to determine whether the key elements of consent were equally understood using the two consent approaches and to assess potential participants’ perceptions about the acceptability of the consent process.
METHODS
Overall Design
We conducted a pilot randomized, controlled trial comparing in-person and videoconference-assisted consent. Due to the nature of the intervention, blinding was not possible. For in-person consent, a physician and a research coordinator were present at the patient’s bedside; for consent by videoconferencing, the coordinator was at the bedside and the physician was present only by real-time videoconferencing.
In order to assess the subjects’ unbiased views of the consent process, we utilized a deception technique in which a sham clinical trial was presented for consideration. The sham trial was a randomized controlled trial comparing two FDA-approved medications for the treatment of hypertension. Once study procedures were completed, the subject was debriefed on the actual intent of the study and provided counselling and information regarding their hypertension. All treatment decisions were at the discretion of the clinical team and were not influenced by the research study. Institutional Review Board (IRB) approval was obtained before any study activity was conducted. The IRB determined that the protocol involved no more than minimal risk and that the deception would have no adverse effects on rights or welfare of the participants with the participants being debriefed after their participation in the trial [9].
Setting
This study took place between March and October 2013, in an urban, academic Emergency Department (ED) with an annual visit volume of ~90,000.
Eligibility and Randomization
Inclusion and exclusion criteria were primarily based on the sham clinical trial. A convenience sample of patients in the ED aged at least 18 years and with two blood pressure readings taken at least five minutes apart that were greater than 140/90mmHg were eligible. A single coordinator screened ED patients for eligibility. Subjects were primarily excluded if they were taking either of the two medications for the sham trial, were eligible for an actual clinical trial, known to be pregnant, non-English speaking, or unable to provide informed consent due to cognitive impairment. We also excluded those with severe illness or pain in order not to interfere with the provision of acute clinical care.
Subjects were randomized to either physician-at-bedside or videoconferencing consent. Sequentially numbered concealed envelopes were used to blind the sequence to the enrolling investigator until enrollment.
Consent for the Sham Trial
The in-person research coordinator screened potentially eligible subjects in the ED, and then introduced the enrolling physician either in person or via videoconference on the iPad. Signed consent forms were not copied into the medical records.
Videoconferencing
Videoconferencing was performed in real-time via Face Time on an AppleiPad2 over Wi-Fi wireless internet using WPA2 enterprise settings and128-bit AES encryption. At the time of enrollment, the research coordinator at bedside introduced the physician (on the iPad), who described study procedures, risks, benefits, voluntariness, conflicts of interest, alternatives and right to withdraw to the subject. The connection provided the necessary security to protect patient information.
Outcomes and Assessments
The primary outcome was the proportion of subjects consenting to the sham clinical trial. We also compared patient comfort (measured on a Likert Scale 0/10 being least comfortable and 10/10 being most comfortable) and knowledge acquisition (measured by yes/no/unsure question format) between each consent technique. All outcomes were assessed by computing the differences in proportions with 95% confidence intervals. All statistical analyses were conducted using SPSS 22.0 (IBM Corporation, Armonk, NY).
The brief questions used to estimate knowledge and understanding of the proposed study are shown below:
Post-Consent Questionnaire:
During the consent process:
- How comfortable were you when talking to the research doctor? ( Scale 0 to 10, where 0 is the least comfortable and 10 being extremely comfortable) _______
- Did the research doctor answer all of your questions about the study?
Yes No
3. Did you have any trouble communicating with the research doctor?
Yes No
If yes, then what were the problems? _______________________________
Regarding the Consent form:
1. Is this study voluntary?
Yes No I don’t know
2. Are there any costs to you for taking part in this study?
Yes No I don’t know
3. Will you be identified by name when we publish this study?
Yes No I don’t know
4. Will you be paid to participate in this research study?
Yes No I don’t know
5. Were there any risks in taking part in this study?
Yes No I don’t know
6. Were you promised any benefits in taking part in this study?
Yes No I don’t know
7. Are you able to withdraw from the study at any time?
Yes No I don’t know
Debriefing by the physician occurred immediately following completion of the acceptability and comfort levels questionnaire where each patient was informed of the true nature of the study and also counselled on their hypertension.
RESULTS
Of 100 patients screened for inclusion, 69 did not meet enrollment criteria for the sham trial and 10 were not interested in participation in clinical research (Figure 1).
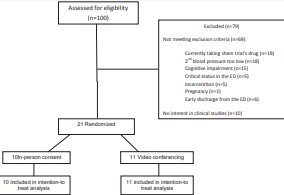
Figure 1 Flow diagram of the strategy used to identify eligible patients
Ten subjects were randomized to consent via the physician-at-bedside and 11 via videoconferencing. Overall, mean age was 43 years (SD 11), 76% were African-American, 62% were male, and 81% had at least a high-school education (Table 1).
|
Table 1: Demographic characteristics by study group. |
||||||
|
Total (N=21) |
Tele-consent (N=11) |
Physician at Bedside (N=10) |
||||
|
Age – Mean (SD) |
43 |
(11) |
44 |
(13) |
42 |
(9) |
|
Race – N (%) |
||||||
|
Black/African American |
16 |
(76.2) |
9 |
(81.8) |
7 |
(70.0) |
|
White |
5 |
(23.8) |
2 |
(18.2) |
3 |
(30.0) |
|
Sex – N (%) |
||||||
|
Male |
13 |
(61.9) |
8 |
(72.7) |
5 |
(50.0) |
|
Female |
8 |
(38.1) |
3 |
(27.3) |
5 |
(50.0) |
|
Insurance Provider – N (%) |
||||||
|
Self-Pay |
11 |
(52.4) |
6 |
(54.5) |
5 |
(50.0) |
|
Medicare |
5 |
(23.8) |
2 |
(18.2) |
3 |
(30.0) |
|
Private |
4 |
(19.0) |
2 |
(18.2) |
2 |
(20.0) |
|
Medicaid |
1 |
(4.8) |
1 |
(9.1) |
0 |
(.0) |
|
*Highest Level of Education – N (%) |
||||||
|
Some HS |
4 |
(19.0) |
2 |
(18.2) |
2 |
(20.0) |
|
HS/GED |
11 |
(52.4) |
6 |
(54.5) |
5 |
(50.0) |
|
Some College |
4 |
(19.0) |
2 |
(18.2) |
2 |
(20.0) |
|
Bachelor’s degree |
2 |
(9.5) |
1 |
(9.1) |
1 |
(10.0) |
|
|
|
|
|
|
|
|
|
Currently use blood pressure medication– N (%) |
4 |
(19.0) |
1 |
(9.1) |
3 |
(30.0) |
|
Abbreviations: *HS: High School; GED: General Education Development Test |
||||||
In the videoconferencing group, 10/11(91%) of participants consented, and in the physician-at-bedside group 8/10 (80%) consented (difference 11%, 95%CI -41%to19%). There were no differences in the acceptability of the use of videoconferencing or in the understanding of key components of the informed consent for the sham clinical trial (Table 2).
|
Table 2: Differences in consent rate, acceptability of consent process, and understanding of key elements of informed consent by study group. Differences between groups and 95% confidence intervals are presented. |
|||||||
|
Tele-consent (N=11) |
Physician at Bedside (N=10) |
Difference |
95% CI |
||||
|
Lower |
Upper |
||||||
|
Patient consented to study – N (%) |
10 |
(90.9) |
8 |
(80.0) |
-0.11 |
-0.41 |
0.19 |
|
*Acceptability of consent process |
|
|
|
|
|
|
|
|
Comfort level – Mean (SD) |
10 |
(1) |
10 |
(1) |
0.23 |
-0.39 |
0.85 |
|
Research doctor answered all questions – N (%) |
10 |
(90.9) |
10 |
(100.0) |
0.09 |
-0.08 |
0.26 |
|
Trouble communicating with research doctor – N (%) |
0 |
(0.0) |
0 |
(.0) |
0.00 |
0.00 |
0.00 |
|
|
|
|
|
|
|
|
|
|
**Understanding of key elements of informed consent – N (%) |
|||||||
|
Is this study voluntary? |
11 |
(100.0) |
10 |
(100.0) |
0.00 |
0.00 |
0.00 |
|
Are there any costs to you for taking part in this study? |
11 |
(100.0) |
9 |
(90.0) |
-0.10 |
-0.29 |
0.09 |
|
Will you be identified by name when we publish this study? |
8 |
(72.7) |
8 |
(80.0) |
0.07 |
-0.29 |
0.43 |
|
Will you be paid to participate in this research study? |
11 |
(100.0) |
9 |
(90.0) |
-0.10 |
-0.29 |
0.09 |
|
Were there any risks in taking part in this study? |
4 |
(36.4) |
3 |
(30.0) |
-0.06 |
-0.47 |
0.34 |
|
Were you promised any benefits in taking part in this study? |
10 |
(90.9) |
9 |
(90.0) |
-0.01 |
-0.26 |
0.24 |
|
Are you able to withdraw from the study at any time? |
10 |
(90.9) |
10 |
(100.0) |
0.09 |
-0.08 |
0.26 |
|
*Acceptability was measured on a 0-10 Likert scale where 0/10 the least comfortable and 10/10 being extremely comfortable. **Understanding of the informed consent was measured by yes/no/unsure question and answer format. |
|||||||
Notably, about a third of patients in both groups incorrectly characterized the risks described by the physician.
DISCUSSION
In this pilot randomized controlled trial, we demonstrated the feasibility of conducting consent for research via video conferencing within a busy urban ED setting. We found that the rate of consent, along with comfort level, was the same in both study arms, and that knowledge or understanding of the proposed trial was similar between the study arms. These findings support the notion that consent into a clinical research study may be obtained successfully via videoconferencing, such as with the iPad.
Prior studies have evaluated the feasibility and reliability of utilizing videoconferencing to augment clinical care in stroke, heart failure, and pre-operative evaluation, but there are no data comparing videoconferencing to in person telemedicine to deliver study information during the informed consent process [2-4].Despite the lack of data, trials are already ongoing using videoconferencing to obtain informed consent. Our pilot data would support this approach. For example, the Antihypertensive Treatment of Acute Cerebral Hemorrhage (ATACH) II trial is an ongoing phase 3 acute stroke treatment trial that allows for the use of existing clinical stroke telemedicine networks for facilitating enrollment of patients [6]. As in our study, ATACH II must have an individual facilitating the consent interaction at the bedside so that the study physician can be introduced and so that the informed consent document can be provided to the patient for signature.
Our study had several limitations. Although the sample size was small, our aims were to determine the feasibility of obtaining consent for a research study with videoconferencing and compare consent rates, subject comfort level, and knowledge transfer between in-person and videoconference consent. Ours is the first study to compare these approaches in an ED. Our urban, predominantly male African American population also limits generalizability. However, it is well recognized that African American males are less likely to participate in research [10]. As such, the high rate of participation we observed is reassuring for the likely participation rates and comfort of other populations with consent via videoconference. Whenever telemedicine is deployed to facilitate research, there must be a means of physically introducing the study, the videoconferencing or telemedicine equipment, and the study physician. There must also be a means for documenting the informed consent, usually by physical signature on a paper document. In many randomized clinical trials, consent is obtained by research assistants, coordinators, and other healthcare providers and not physicians. The use of a research coordinator at bedside to facilitate this may have influenced the willingness of subjects to participate. While all consent procedures were conducted by the physician, it is unclear if consent rates would have differed if only clinical personnel were involved in the physical interaction with patients. We devised a deceptive sham clinical trial in order to blind subjects to the actual intervention of in-person versus videoconference consent. Although ethically controversial, minimal risk was posed to participating subjects, IRB approval was obtained prior to conducting any study procedures, and subjects were fully debriefed following their participation [11]. Further, given the minimal risk posed by our sham trial of approved medications, we may have overestimated comfort with the consent process.
In this small randomized trial, we demonstrated that obtaining informed consent via videoconference resulted in no difference in patient comfort and knowledge of study procedures compared with in-person physician consent [12]. Larger studies to demonstrate the equivalence of these consent approaches are warranted in order to inform current regulations and to provide guidance on linking the physical and video components of the process.









































































































































{kind=link}