Parkinson’s Disease: The Catabolic Theory
- 1. University College London, Institute of Neurology, WC1N 3BG, Queen Square, London, UK
Citation
Manzoni C (2013) Parkinson’s Disease: The Catabolic Theory. J Neurol Transl Neurosci 1: 1009.
Editorial
Parkinson’s disease (PD) is one of the most studied neurodegenerative disorders. In the majority of cases, PD shows no obvious genetic cause and it is classified as idiopathic or sporadic disease. This does not mean that there is no genetic involvement; in fact, genome wide association studies (GWAs) have highlighted the existence of multiple risk loci all over the genome able to increase an individual’s propensity to develop sporadic PD. In light of this, sporadic cases may be considered the result of a predisposed genotype stimulated by the correct environmental exposure.
As for many other neurodegenerative disorders, there is a small but significant minority of PD cases that display classical, Mendelian patterns of inheritance (familial PD, fPD) caused by genetic mutations in single loci that, despite variable degrees of penetrance, segregate with the disease. In addition to fPD genes, there is also a host of mutations in genes associated with parkinsonism, a clinical syndrome that is not classified as pure PD despite some overlaps in terms of clinical and/or pathological presentation.
Familial cases provide an intriguing and potentially high informative, paradigm for neurological diseases, even if they represent a minority of cases. In patients with familial disease, the molecular cause of the pathology is very well defined although its action may not be fully understood. There is an implication in research following on from this observation. We use genetic cases to dissect out the mechanism relating the mutation in protein X to the pathology Y. The final aim of this approach is the elucidation of a molecular mechanism that can be extrapolated from the familial cases to cast light on the pathogenic event triggering the more common, but also more difficult to study, sporadic form of disease. There is growing evidence that this approach is valid; i. e. the case of Alzheimer’s disease (AD) where the study of familial AD indicated the Aβ peptide as the pathological culprit in both familial and sporadic cases, culminating with the construction of the amyloid cascade hypothesis [1]. Many studies have now been carried out to investigate fPDs and parkinsonisms, so in the coming years we may hope to assist to such a breakthrough for PD as it has been for AD.
Active research is now on going for the protein products of genes involved in fPD and parkinsonism. Leucine rich repeat kinase 2 (LRRK2) is an enzyme (kinase and GTPase functions coexist in the same open reading frame) mutated in the major number of familial PD cases [2,3]. Recently, the potential involvement of LRRK2 in autophagy has become the focus of a growing body of literature. Manipulating the kinase activity of LRRK2, protein over-expression or knock-down; all modify autophagy [4,6]. Fibroblasts from patients carrying LRRK2 mutations [7] as well as dopaminergic neurons derived from iPS cells of LRRK2 patients [8] show autophagic alterations. Transgenic mice expressing mutant LRRK2 have alterations of autophagy in the brain [9] while knock-out mice, despite the absence of any obvious neuronal phenotype, show alterations of autophagy and lysosomal functionality in the kidneys [10].
Glucocerebrosidase (GBA) is a lysosomal enzyme responsible for the catabolism of glucocerebroside releasing glucose and ceramide; homozygous mutations in GBA cause Gaucher’s disease (a lysosonal storage disorder). Within the heterozygous carriers there is increased incidence of PD due to an as-yet unclear aetiology. Heterozygous mutations in GBA may cause a loss of lysosomal function too mild to result in Gaucher’s disease, but strong enough to reduce autophagy leading to an accumulation of misfolded proteins like αsynuclein [11]. Alternatively, mutant GBA can misfold and engulf the ubiquitin-proteosome system [12]. Independent of mechanism, GBA mutations result in a global reduction in the cell catabolic payload. On the top of that, since ceramide is known to regulate autophagy [13], the reduction in its production due to GBA mutations may be enough to alter the catabolic cell homeostasis.
The accumulation of αsynuclein in Lewy’ bodies, is one of the key hallmarks of PD. There are mutations in αsynuclein that associate with familial PD which are thought to increase the ability of the protein to aggregate in amyloid deposits. Mutant forms of αsynuclein however, are proven to inhibit autophagy by mediating a direct block of lysosomal uptake/functionality [14].
Mutations in ATP13A2 associate with both lysosomal disorders and a parkinsonian’s syndrome named Kufor-Rakeb [15], they cause the ATP13A2 protein to misfold and undergo premature degradation [16] ending in accumulation of impaired lysosomes and undigested autophagosomes [17]. A similar scenario is descriptive for mutations in VPS35, a component of the retrotransport shuttle that distributes receptors for hydrolytic enzymes between ER and lysosomes. Mutations in VPS35 associate with parkinsonism and they cause impairment in lysosomal functionality [18-19].
Mutations in PINK1 and parkin are found in early onset parkinsonism. These two proteins are differently involved in the regulation of mitochondrial fission and fusion, biogenesis and mitophagy [20]. As an overall, their physiological activity is responsible to regulate the mitochondria quality control and to maintain the correct pool of functional mitochondria within the cell. A less studied protein involved in early onset parkinsonism is DJ-1; among other actions, DJ-1 has been preliminarily reported in oxidative stress and mitochondrial homeostasis [21].
Each of the listed genes has its own character and when mutated in pathology, originates a peculiar spectrum of molecular alterations that culminates in a specific clinical phenotype. However, in the morass of molecular alterations, there is a recurrent theme: the impairment of catabolic processes and degradation pathways. Autophagy, mitophagy, lysosomal or proteosomal activities are commonly impaired regardless the specific gene under analyses (Figure 1)
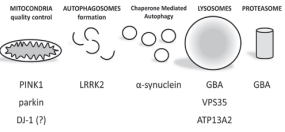
Figure 1 Putative role of parkinsonian proteins in catabolic processes.
and they are slowly emerging in the scientific debate as a possible unifying scenario in familial cases [22,23]. Can alterations in degradation pathways be the clue that we are hunting for in familial cases to cast a light onto the pathogenesis of sporadic PD?
For amyloid pathologies there is a general acceptance that the accumulation of misfolded proteins can follow an over-production of the protein itself, a hyper-aggregation but also a global reduction in clearance mechanisms. Thus, the accumulation of αsynuclein in sporadic PD could be considered as a proof of concept for a pathogenic mechanism centered on catabolic alterations. From a more mechanistic point of view, hsp70, implicated in chaperone mediated autophagy, and LAMP2A both show decreased expression in post mortem PD brains [24] with a consequent possible reduction in autophagic activity. The demonstration of catabolic alterations in sporadic PD is however far from achieved and more in-depth studies are necessary. We also need to determine how and why a general alteration in cell catabolism generates damage to the brain only. It may be speculated that neurons, as non-replicating cells, are more susceptible than any other cell type to alterations in degradation pathways because they cannot dilute any waste by cell division, and they cannot fix any damage by simply being replaced. But then, with the entire brain available, we need an explanation of why PD degeneration, after following a conserved spreading route, is so aggressive in the substantia nigra pars compacta.
Can a catabolic-only hypothesis be descriptive of the pathogenesis of PD in the same fashion that the amyloid-only hypothesis has been for AD? To answer this question there is a requirement to move from the study of genetic forms of the pathology to sporadic cases. In the context of sporadic disease, catabolic alterations may not be present or they can be a consequence of chronic illness rather than the cause of disease. This highlights the urgent need to develop systems which will allow us to test whether the disruptions in catabolic pathways that have been linked to fPD are also observed, and in a causative manner, in idiopathic PD.
We are still far from a final answer as to what underlies the pathological process in PD, but the clues provided by studying familial forms of PD give us hope that we are at least some way along the journey to a level of understanding that will allow us to intervene and alter the course of this devastating disorder.
ACKNOWLEDGEMENTS
Thanks to Dr. PA Lewis for critical comments and reading of the manuscript. CM is supported by the Rosetrees Trust and acknowledges generous funding from the Michael J. Fox Foundation. This work was supported in part by the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson’s Disease Consortium (UKPDC) whose members are from the UCL Institute of Neurology, the University of Sheffield and the MRC Protein Phosphorylation Unit at the University of Dundee.







































































































































{kind=link}