The EP3 Receptor: Exploring a New Target for Type 2 Diabetes Therapeutics
- 1. Department of Nutritional Sciences, University of Wisconsin-Madison, Wisconsin, USA
- 2. Department of Medicine, University of Wisconsin-Madison, Wisconsin, USA
Citation
Neuman JC, Kimple ME (2013) The EP3 Receptor: Exploring a New Target for Type 2 Diabetes Therapeutics. J Endocrinol Diabetes Obes 1(1): 1002.
Editorial
The number of Americans diagnosed with type 2 diabetes in 2012 was predicted by the American Diabetes Association (ADA) to be 23.3 million, with a projected cost of $306 billion: more than $1 for every $5 spent on healthcare [1]. While many type 2 diabetes therapies exist, non-adherence and ineffective daily blood glucose management promote associated complications. Most classic type 2 diabetes therapies aim to reduce peripheral insulin resistance, whereas more recently-developed therapeutics aim to improve beta-cell function by acting directly on the insulin-producing cells of the pancreatic islet. Furthermore, beta-cell therapies that work through G protein-coupled receptors modulate insulin secretion in response to stimulatory glucose, thus promoting a more physiological regulation of the insulin response. Examples of these therapeutics are the stable glucagon-like peptide 1 (GLP-1) agonists or dipeptidyl peptidase 4 (DPP-4) inhibitors, which both act through the GLP-1 receptor on the beta-cell to stimulate downstream signaling pathways (Figure 1).
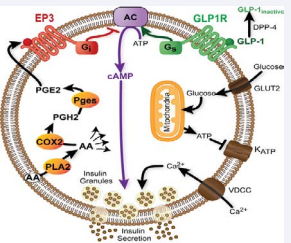
Figure 1 PGE2 signaling through EP3 negatively regulates beta-cell function. AA is cleaved from membrane phospholipids by phospholipase A2 (PLA2). PGE2 is generated by two sequential enzymatic steps: cyclooxygenase 2 (COX2)-mediated generation of PGH2, followed by prostaglandin E synthase (Pges)-mediated conversion to PGE2. PGE2 can activate the EP3 receptor on the beta-cell to block cAMP production, interfering with cAMP-mediated signaling through the GLP-1 receptor.
An emerging target, the E-prostanoid receptor 3 (EP3), plays a critical regulatory role in modulating beta-cell function and may be a key component in the development of beta-cell pathologies. It is well established that the endogenous ligand for EP3, prostaglandin E2 (PGE2), negatively regulates insulin secretion [2-4]. PGE2 is rapidly degraded in the bloodstream and is thought to act on its target tissues via autocrine or paracrine mechanisms [5]. Increased (1) plasma levels of PGE2 metabolites, (2) production of PGE2 from platelets and islet cells (3) expression of PGE2 synthetic enzymes, and (4) EP3 expression have all been linked with the pathophysiology of type 2 diabetes [6-10]. Furthermore, increased islet PGE2 production and EP3 expression have a negative impact on signaling through the GLP-1 receptor, which in beta-cells is a strong, endogenous potentiator of insulin secretion in response to food intake [6] (Figure 1). Increased EP3 signaling might explain the failure of type 2 diabetes drugs that act through the GLP-1 receptor in a subset of type 2 diabetic patients [6-11]. Although the EP3 receptor plays critical roles in islet biology, extrapancreatic function may be equally important in both type 2 diabetes pathophysiology. PGE2 signaling through EP3 appears to be a critical component of reducing lipolysis in adipose tissue. Mice lacking a critical phospholipase A2 , an essential initiating component in PGE2 biosynthesis, have reduced PGE2 concentrations in adipose tissue and are resistant to weight gain [12]. Although these mice are protected from weight gain and present an increased catabolic phenotype, they are largely insulin resistant in the liver and have lowered peripheral glucose metabolism, most likely due in part to reduced fat mass [12]. In addition, EP3 signaling can mediate the migratory response of vascular smooth muscle cells (VSMCs). Pharmaceutical blockade of EP3 or its genetic deletion produces VSMCs that exhibit suppressed G protein-mediated signaling and altered polarity and directional migration, suggesting that blockade of EP3 might protect from dysfunctional vascular remodeling [13]. Furthermore, EP3-null mice exhibit reduced baseline mean arterial pressures, and pharmacological inhibition of EP3 blocks angiotensin 2-mediated vasoconstriction, suggesting the EP3 receptor as a target for antihypertensives [14]. Thus, it appears that PGE2 signaling through EP3 may play a significant role in the progression of obesity, diabetes, and co-morbidities such as cardiovascular disease.
Even though PGE2 signaling through EP3 may be important in metabolic disease progression, EP3 has other functions that lend caution to EP3 as a therapeutic target. First, signaling through EP3 reduces cancer cell proliferation and tumorigenesis through G12-mediated Rho activation [15]. Blockade of EP3 also prevents apoptosis, which protects from stroke injury [16], but could potentially interfere with cancer cell killing. Extending from these findings, caution should be taken to ensure that pharmaceutical blockade of EP3 does not promote tumorigenesis in vivo. In addition, whole-body EP3-null mice develop obesity, insulin resistance, glucose intolerance, and eat considerably more than wild-type controls [17]. Part of this phenotype can be explained by increased night eating, most likely due to unstable sleep patterns, as PGE2 (acting through hypothalamic EP3) may act as a somnogen [18]. Furthermore, high levels of PGE2 and increased signaling through EP3 augment nitric oxide synthase expression, an enzyme critical in the brain development of newborns [19]. This signaling cascade is suggested to play a significant role in connecting brain circulation and synaptic activity in perinatal development. A drug that did not cross the blood/brain barrier might bypass these negative consequences of inactivation of brain EP3 signaling. Another way to target a GPCR is to interfere with its downstream signaling mechanisms. EP3 is one of four known E-prostanoid receptors (EP1-4) and is widely expressed across tissues, with highest expression in the kidney, uterus, and pancreas [20]. When bound by PGE2, EP3 contributes to a reduction in intracellular cyclic AMP (cAMP) concentrations by inhibiting adenylyl cyclase activity [21,22] (Figure 1). A unique aspect of EP3 is that multiple splice variants exist in every species tested, differing only in their C-terminus (Table 1).
Table 1: Human, Mouse, and Rat EP3 splice variants, organized based on unique C-terminal sequence and arranged with their species homologs. The C-terminus of these splice variants appears to impact on G protein coupling, constitutive activity, and desensitization to agonist. The G proteins with the most evidence for coupling are shown in Bold.
| Species | Variant | Unique C-terminal Sequence | G-prot. coupling | Ref. | Constitutive activity | Ref. | De-sensitized? | Ref. |
| Hs | Var 4 | IRYHTNNYASSSTSLPCQCSSTLMWSDHLER | Gi, Gq, Gβγ | [20,26-28] | None (Gi) | [29] | ||
| Mm | alpha | IRDHTNYASSSTS_LPCPGSSALMWSDQLER | Gi, G12, Gβγ | [30-32] | Partial (Gi) Full (G12) | [30, 33] | Yes-Slow & persistent | [34] |
| Rn | A | IRDHTNYASSSTS_LPCPGSSVLMWSDQLER | Gi | [35] | Full (Gi) | [35] | Yes | [36] |
| Hs | Var 5 | VANAVSSCSNDGQKGQPISLSNEIIQTEA | Gi, Gs, Gq, Gβγ | [20,26,27,37] | None (Gi) | [29] | Yes-Slow & persistent | [37] |
| Mm | gamma | VANAVSSCSSDGQKGQAISLSNEVVQPGP | Gi, Gs, Gβγ | [31,34,38] | Full (Gi), None (Gs) | [39] | ||
| Rn | B | VANAVSSCSSDQQKGQIASLSNEVVHPGP | ||||||
| Hs | Var 6 | MRKRRLREQEEFWGN | Gi, Gq, Gσ | [20,26,27,37] | Partial (Gi) | [29] | Yes-Rapid & transient | [37] |
| Hs | Var 7 | EEFWGN | Gi, Gq, Gβγ | [20,26,27,37] | Full (Gi) | [29] | Yes-Rapid & transient | [37] |
| Hs | Var 8 | MRKRRLREQLICSLQNSQIQRATAHCGQVQTYRVLNREEMEVLVSSINVYTRISTVKTE | Gi | [20,27] | ||||
| Mm | Beta | MMNNLKWTFIAVPVSLGLRISSPREG | Gi, Gβγ, G12 | [30-32] | None (Gi), None (G12) | [30, 33] | None | [34] |
| Rn | Beta | MMNNLKRSFIAIPASLSMRISSPREG | None | [36] | ||||
| Rn | D | FSLCFNR |
The identity of the C-terminal tail appears to determine which G protein-mediated signaling cascades will be activated, as well as whether the receptor exhibits constitutive activity or the ability to be desensitized by agonist. Of note, in the pancreatic beta-cell, the EP3 receptor can couple to a unique Gi subfamily member, Gz [21,23]. Gz is the most biochemically-unique and tissue-restricted Gi -subfamily G protein [24,25]. Determining which splice variant is responsible for Gz -coupling in the beta-cell may lead to new ways to specifically target this interaction. In sum, we have learned much about the role of PGE2-mediated EP3 signaling in metabolic diseases such as type 2 diabetes, yet we still have a long way to go before confirming that EP3 is a suitable target for new type 2 diabetes therapeutics. Overall, though, the results seem promising.







































































































































{kind=link}