Attenuation of Cellular and Molecular Hallmarks of Aging by Physical Exercise Intervention
- 1. Key Laboratory of Cellular Physiology, Shanxi Medical University, South XinJian Road 56, Taiyuan, Shanxi, PR China
Abstract
Secondary aging is characterized by nine cellular and molecular hallmarks including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, Stem Cell (SCs) exhaustion, and altered intercellular communication. Exercise might be a uniquely valid intervention in promoting healthy aging and attenuating aged-related diseases without any side effects. On the molecular mechanism, exercise can effectively arrest all of the nine hallmarks of aging mentioned above. This review describes the mechanisms by which exercise can ameliorate these processes of aging.
KEYWORDS
- Aging
- Genes
- Telomere
- Epigenetics
- Proteostasis
- Nutrient Sensing
- Mitochondria
- Cellular Senescence
- Stem Cell
- Intercellular Communication
CITATION
Li L (2024) Attenuation of Cellular and Molecular Hallmarks of Aging by Physical Exercise Intervention. J Family Med Community Health 11(2): 1204.
BACKGROUND
It is well known that no therapeutic strategy for chronic diseases is better than primary prevention and no strategy for prolongation of lifespan is better than earlier prevention. As one of the parameters considered of primary prevention, correct lifestyle, i.e., physical activity and nutritional balance is the crucial strategy. Aging is characterized by the decline of systems and organs functions. Exercise is the best strategy for preventing against the decline of systems and organs functions following aging. Aging is a very complex physiological process controlled in part by genetic programs. Several studies evidenced that reduction of exercise and losing of independence in later life was closely related to genetic alteration [1].
In prehistoric times, superior sport ability of human was needed for survival and evolution. Today, two polarizations for sport ability of human are apparent. In the arena, athletes run faster, jump higher. In daily life, more and more people lose their sporting ability. More and more people who have lost their fitness have lost their good health, too. Fatigue with aging signifies the declining of the individual’s physical and mental resources and is considered as a characteristic of aging [2]. Exercise is considered as a powerful intervention for delaying aging and preventing fatigue based on the physiological effects [3]. On the other hand, the main pathogenesis of chronic disease is attributed to dysmetabolism. A lack of physical activity is an extremely dangerous behavior for most patients who have suffered from dysmetabolism and chronic diseases.
In general, aging is characterized by the accumulation of damage and other deleterious changes, leading to the loss of functionality and fitness. As an external factor, exercise represents a major challenge to whole-body homeostasis (internal factor). For meeting this challenge, acute and adaptive responses in systemic and organic levels i.e., physiological adaption, are taking place in our body to keep homeostasis. For meeting this challenge, some changes in molecular and genetic level are taking place to retard aging. As an inevitable program of life aging is characterized by a time-dependent progressive loss of the individual’s physiological function and gradual increase of aged-related diseases and mortality [4]. Considering the complex interactions between genetic predisposition and environmental factors, it is an enormous challenge to delay aging and prevent aging-related diseases [5].
Based on modern biomedical knowledge, aging is regarded as a programmed process which is controlled by genetic programming and cumulative damage of cells [6]. The former is regarded as an intrinsic biologic programmed deterioration of cells following genetic programming (e.g. primary aging). The latter is regarded as the cumulative damage in cells and organs aggravated by extrinsic sources (e.g. secondary aging). Primary aging corresponds to the progressive and inevitable decline in cellular structure and physiologic function that happens independently of lifestyle, environmental influences, or disease. Untoward change involving interactions of primary aging with environmental influences and disease is defined secondary aging. Seals, et al. [7] proposed that optimization of physiological function and/or maintenance of organ reserve capacity throughout the lifespan should be an aim to delay lifespan and improve life quality. They defined effective strategies to maintain organ reserve capacity and/or abolish decline of physiological function with aging as primary prevention and improve or slow further decline of physiological function and organ reserve capacity in older people with already impaired function as secondary prevention. Exercise is the best effective strategy to realize the aim due to its unique effect in improving multi-organ functions, inducing metabolic demand for more ATP production, costing more energy to reduce bodyweight.
Summarizing present knowledge of molecular biology in aging López-Otín, et al. [4] proposed nine cellular and molecular hallmarks that contribute to the process of aging, including genomic instability; telomere attrition; epigenetic alterations; loss of proteostasis; deregulated nutrient sensing; mitochondrial dysfunction; cellular senescence; SCs exhaustion; and altered intercellular communication. Basic physiological change in exercise is acceleration of metabolism to compensate the deficit from ATP consumption induced by muscle contraction. For producing more ATP and ensuring accelerating metabolism, the heart has to transport more blood by enhancing contraction; lungs have to breathe in more oxygen and breathe out more carbon dioxide by enhancing constriction of respiratory muscles; the liver and kidney have to detoxify and discharge more toxin produced by accelerating metabolism; and the neuroendocrine system is involved in regulating organs activities etc. As results, physiological functions of all organs are improved and all organs keep a younger status. In other words, exercise is a response of body to environment change (metabolism enhancement). The stronger (or younger) functions of organs we have, the better suitability for environment changes we get.
Decline of physiological functions in various organs and/ or systems is a decline in whole body level. Almost all of the declines are related to nine cellular and molecular hallmarks in aging. Almost all of nine cellular and molecular hallmarks in aging are involved in various aged-related diseases. Exercise enhances physiological functions and prevent against aged-related diseases. On the molecular mechanism level, exercise attenuates all of nine cellular and molecular hallmarks in aging. In present review significances of exercise in protecting against the nine cellular and molecular events of secondary aging were reviewed.
METHODS
Data Sources
The literatures in PubMed were scanned using a combination of keywords involved in secondary aging molecular hallmarks including telomere; epigenetics; proteostasis; nutrient sensing mitochondria; cellular senescence; stem cell; intercellular communication, and sports; aging; genes for peer-reviewed. Further, relevant papers that were listed as references in the initial scanning papers were selected. The present review is a narrative review rather than a systematic review, it did not strictly follow the guidelines for ‘‘Preferred Reporting Items for Systematic Reviews and Meta-Analyses’’ (PRISMA; http://www. prisma-statement.org/).
Study Selection: The criteria for article inclusion were (1) original research or reviews in PubMed, (2) written in English, (3) published in 2002 or later.
Data Extraction
The literatures was reviewed and positive evidence to show the relevance between secondary aging molecular hallmarks (telomere; epigenetics; proteostasis; nutrient sensing mitochondria; cellular senescence; stem cell; intercellular communication) and sport were used in the present review.
FINDINGS AND DISCUSSION
Rebelo-Marques, et al. [6] overall reviewed benefit of exercise in attenuating hallmarks of aging. Many studies reveal the enormous potential obtained by exercise in maintaining genomic stability [8]; attenuating telomere attrition [9]; preventing epigenetic alterations [10]; maintaining proteostasis [11]; regulating nutrient sensing [12] enhancing the mitochondrial function [13]; delaying cellular senescence [14]; reducing SCs exhaustion [15]; altering intercellular Communication [16]. (Table1, Figure 1).
Table 1: Effects of exercise on nine hallmarks in secondary aging.
|
Variable (nine hallmarks in secondary aging) |
Observation (effects of exercise) |
References |
|
Chromosomal instability |
Reduce nuclear DNA damage and repair the damage. |
[17-22] |
|
Increase levels of BDNF, activates CREB, and upregulate APE1. |
||
|
Protect mtDNA stability, reduce multisystem pathology and prevent premature mortality |
||
|
Attenuate 8-OHdG content. |
||
|
Prevent increase of genomic oxidative DNA damage and improve the genetic stability. |
||
|
Telomere attrition |
Maintain telomere length. |
[23-28] |
|
Decreas expression of p53 and shelterin complex. |
||
|
Increase mRNA levels of DNA repair enzymes Ku70 and Ku80 and mRNA levels of TRF1, TRF2, and Pot-1. |
||
|
Upregulate TERTmRNA and sirtuin-6 mRNA. Regulate fifty-six miRNAs |
||
|
Upregulate four miRNAs (miR-186, miR-181, miR-15a and miR-96). |
||
|
Epigenetic inadaptation |
Regulate epigenetic modification. |
[29-32] |
|
Regulate DNA methylation in 60% of imprinted loci including RB1, MEG3, UBE3A, PLAGL1, SGCE, INS. |
||
|
Increase lysine 36 which is associated with transcriptional elongation. |
||
|
Induce the export of HDAC 4 and 5 from the nucleus. |
||
|
Activate the AMP-activated protein kinase and the calcium-calmodulin-dependent protein kinase II. |
||
|
Nutrient sensing imbalance |
Regulate nutrient sensing and maintains proteostasis. |
[33-36] |
|
Regulate cellular growth and protein synthesis. |
||
|
Enhance autophagy and inhibits mTOR pathway. |
||
|
Improve abnormal mTOR and phosphorylation of PI3K/AKT proteins and inhibit its activity via increased GSK-3β phosphorylation. |
||
|
Increase the expression of Beclin-1 protein. Reverse hyperphosphorylation and aggregation of tau phosphorylated at the site of Ser199/202, Ser404, Thr231, PHF-1. |
||
|
Increase several molecules which are involved in the mTOR pathway including BDNF, phosphorylated and total mTOR, phosphorylated and total S6 protein and inhibit elongation factor 4E-BP2 |
||
|
Impaired proteostasis |
Attenuate anabolic resistance. |
[37-57] |
|
Produce more energy by promoting catabolism of muscle glycogen and promote muscle growth by increasing anabolism of protein. |
||
|
Keep proteostasis in muscles stimulated by promoting anabolism of muscles protein and inhibiting catabolism of muscles protein. |
||
|
Promote dietary amino acid ingestion and enhance post-exercise rates of muscle protein synthesis. |
||
|
Inhibit nutrient receptor, in turn, speeds catabolism of glucose and fatty acid. |
||
|
Induce significant increase of endogenous hormonal T, GH, IGF-1 elevations. |
||
|
Enhance protein synthesis, decrease protein breakdown, activate satellite cells, enhance Wnt signaling, and promote sex hormone- binding globulin receptor binding. |
||
|
Increase intracellular calcium. |
||
|
Elevate GH, IGF-1, testosterone, or sex hormone-binding globulin. |
||
|
Reverse the changes of several biomarkers of muscle protein synthesis, e.g. increasing protein levels of the unfolded protein response markers (GRP78, DERLIN-1 and CHOP), accumulating misfolded and polyubiquitinated proteins, and reducing chymotrypsin-like proteasome activity. |
||
|
Activate autophagy and re-establish proteostasis. |
||
|
Prevent autophagic flux from skeletal muscle and loss of proteostasis. |
||
|
Increase circulating anabolic hormones (e.g., GH, IGF-1, free IGF-1, testosterone, free testosterone, dehydroepiandrosterone, dihydrotestosterone, and luteinizing hormone) |
||
|
Impaired mitochondrial functions |
Promote robust mitochondrial adaptations by increasing mitochondrial in number and size. |
[58-68] |
|
Enhance Ca2+ release from the sarcoplasmic reticulum and enhancing muscle force, as well as the generation of ATP from respiring mitochondria. |
||
|
Activate the signalling kinases and increasing the expression of mtDNA transcription factor. |
||
|
Activate the UPRmt and further upregulating transcription of mitochondrial chaperones and proteases and equipping the organelle with an augmented capacity for protein folding. |
||
|
Enhance expression of mitochondrial fusion proteins Mfn1/2 along with Opa1 and further facilitating the fusion of the outer and inner membranes, and improve of metabolites of neighbouring organelles. |
||
|
Enhance mitophagy to clean out the damaged organelles and activate lysosome to degrade the dysfunctional mitochondria and release the constituent amino acids for cellular recycling. |
||
|
Increase PGC-1α signalling for mitochondrial biogenesis and fission. |
||
|
Improve aerobic capacity and skeletal muscle mitochondrial respiration. |
||
|
Increase gene transcripts and protein synthesis of mitochondria. |
||
|
Reduce mitophagy flux, i.e., accumulation of autophagy-related markers. |
||
|
Reduce mitochondrial number:size ratio, and Increased mitochondrial bioenergetics. |
||
|
Increase Cisd 1 and 2 as well as mitochondrial protein expression. |
||
|
Cellular senescence |
Attenuates markers of senescence, including p16, EGFP, senescence-associated β-galactosidase, and SASP. |
[69-75] |
|
Decreases expression of transcription factors related to cellular senescence including p53, CDKI, p21 and p16, lipofuscin, TNF-α and IL-1β, and markers of oxidative stress (TNF receptor-associated factor 6, c-Jun N-terminal kinase, and c-Jun). |
||
|
Increases the CD4+/CD8+ and CD4+/CD3+ ratio and decrease of the CD8+/CD3+ ratio. Increase CD4+/CCR7+/CD45RO+ central memory cells, naive CD8+/CCR7+/CD45RO- and CD8+/CCR7+/CD45RO+ central memory cells, and decrease CD4+/CCR7-/CD45RO- TEMRA cells, CD8+/CCR7-/CD45RO- TEMRA cells and CD16+ cells. |
||
|
SCs exhaustion |
Slows ASCs depletion. |
[76-108] |
|
Promotes proliferation of ASCs and increase circulating haematopoietic stem and progenitor cells. |
||
|
Reduces phosphoinositide 3-kinase mediated apoptosis. |
||
|
Increases biomarkers of ASCs including PGE1, vascular VEGF, and SDF 1. |
||
|
Facilitates MSCs migration by increasing IL-6 and recruit SCs. |
||
|
Promotes differentiation of MSCs into osteoblasts and impede differentiation into adipocytes by increases expression of Runx2, Osx, and I-collagen protein, and inhibits expression of PPARγ-2 and C/EBPα. |
||
|
Activation of the satellite cells by increase the expression of VEGF, IGF, NO and hepatocyte GF, fibroblast GF. |
||
|
Promotes hepatocyte growth factor, hepatocyte growth factor activator, hepatocyte growth factor activator inhibitor-1, and hepatocyte growth factor activator inhibitor-2. |
||
|
Diminishes leptin production in adipose tissue, and augment quiescence-promoting hematopoietic niche factors in leptin-receptor- positive stromal bone marrow cells. |
||
|
Increases VEGF expression, in turn, triggers release of haematopoietic and endothelial progenitor cells. |
||
|
Enhances VEGF, IGF-1 concentration and promotes ASCs proliferation. |
||
|
Promotes proliferation and differentiation of neuronal SCs induced by BDNF and satellite cell proliferation mediated by LIF and PDGF. |
||
|
Abnormal intercellular communication |
Attenuates cell cycle inhibitors including p21CIP, p53, and p16INK4A |
[109-120] |
|
Inhibits the exosomes which 1) release during senescence of fibroblast, epithelial cells, and cancer cells; 2) increases with aging; and 3) changes composition (miRNAs, proteins, or lipids) with aging. |
||
|
Promotes release exosome markers (CD63, Alix, CD81, and Flot-1). |
||
|
Promotes proliferation and differentiation of skeletal muscle by increasing release of exosome-carried miRNAs, including miR-1, miR-133a, miR-133b, miR-206, and miR-486. |
||
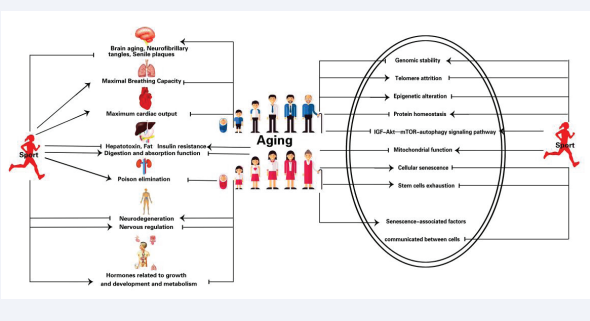
Figure:
Exercise Protects Chromosomal Stability
There is also compelling evidence suggesting that correct life-style i.e., regular physical activity, a well-balanced diet and minimized psychosocial stress can reduce DNA damage and improve chromosomal stability. Constant cytotoxic and genotoxic aggression is considered to be the critical aging hallmark. The accumulation of unrepaired damaged DNA, damages of DNA integrity, and genome instability are crime culprits to induce cellular death and speed aging [18,19]. Exercise can effectively reduce nuclear DNA damage and is regarded as a potential strategy for further DNA damage repair [20].
Brain-Derived Neurotrophic Factor (BDNF) may be a key molecule for NDA repair in brain [21]. BDNF binds to the tropomyosin receptor kinase B receptor and activates intracellular signaling pathways [21]. In addition, BDNF performs its protective effects in neuronal DNA repair via activating cAMP response element-binding protein (CREB). The latter mediates expression of apurinic/apyrimidinic endonuclease 1 (APE1) which is a key enzyme for base excision in the DNA repair pathway. Vilela [17] showed that treadmill exercise promoted spatial memory in aging rats through BDNF signaling, which regulates proteins of the glutamatergic system. Yang, et al. [20] showed that running wheel exercise significantly increased levels of BDNF, activated CREB, and upregulated APE1 in the cerebral cortex and hippocampus of mice. Mitochondrial DNA (mtDNA) mutator mice in which aging phenotypes are speedily expressed are extensively utilized to explore damage of DNA repair in aging. Safdar, et al. [22] showed that a 5-month aerobic exercise program protected mtDNA stability in multiple tissues of mtDNA mutator (progeroid) mice, thereby reducing multisystem pathology and preventing premature mortality.
Modifications of 8-hydroxy-2’-deoxyguanosine (8-OHdG), which is involved in the interaction between •OH or •O2 and G of the DNA strand, is regarded as a biomarker for DNA repair.
To explore effects of aerobic exercise in improving DNA repair mechanisms (e.g., proteasome complex), Radak, et al. [23] measured 8-OHdG content in running training rats. The results showed that treadmill running for eight weeks resulted in nearly a 40% increase in maximal oxygen uptake in both middle-aged (20-month-old) and aged (30-month-old) rats. The age-associated increase in 8-OHdG content was significantly attenuated in the gastrocnemius muscle by exercise. The 8-OHdG repair, which was measured by the excision of 32P-labeled damaged oligonucleotide, increased in muscle of exercising animals. The reactive carbonyl derivatives (RCD) of proteins did not increase with aging. However, when the muscle homogenate was exposed to a mixture of 1 mM iron sulfate and 50 mM ascorbic acid, the muscle of old control animals accumulated more RCD than that of the trained or adult groups. The chymotrypsin-like activity of the proteasome complex increased in muscle of old trained rats. The study suggests that regular exercise-induced adaptation attenuates the age-associated increase in 8-OHdG levels, and increases DNA repair and resistance against oxidative stress in proteins.
Dimauro, et al. [24] analysed contrastively the effect of exercise on spontaneous and H2O2-induced DNA damage and on the transcriptional level of genes involved in DNA repairs systems in diabetes subjects trained and untrained. They found that physical activity to prevent significant increase of genomic oxidative DNA damage induced by chronic exposure to pro- oxidant stimulus and prolonged exercise training improved the genetic stability and the functionality of specific genes.
Exercise Protects from Telomere Attrition
Telomeres are G-rich repetitive DNA sequences located at distal ends of chromosomes and safeguard genetic integrity. As a molecular clock, telomere shortening cue to cellular senescence. Abnormally short telomeres may contribute to aging and aged- related diseases. Inactive lifestyle which is a key culprit leading aging and age-related metabolic diseases including obesity, type 2 diabetes mellitus, chronic cardiovascular disease, dementia, osteoporosis, and cancers could be attributed to short leukocyte telomeres. Exercise promotes healthy aging and protects from age-related diseases whilst concurrently maintaining telomere length. The positive relationship between exercise and telomere length raises the possibility of a mediating role of telomeres in retarding aging and preventing aged-related diseases via exercise [25].
Epidemiologic surveys showed that exercise is a powerful strategy for maintenance of longer telomere length. Muniesa, et al [26] evidenced that athletes have longer telomeres than controls after comparing telomere length between young athletes and healthy nonsmokers, physically inactive controls. Similarly, Simoes, et al. [27] showed that sprinters had longer telomere length, lower body fat and BMI, and a better lipid profile than controls after comparing telomere length of sprinters and controls. Cunha, et al. [28] evaluated the relationship between exercise intensity (moderate vs. high-intensity) and telomere length and senescence markers including checkpoint kinase 2, shelterin telomere repeat binding 1 and 2 (Trf1/Trf2), tumor suppressors (Chk2 and p53,), DNA repair (Xrcc5), telomerase reverse transcriptase (mTERT) in middle aged mice which were grouped as controls, swimming mice. After training for 12 weeks, a longer telomere length, decreasing expression of p53 and shelterin complex were shown in the high-intensity exercise mice than that of the low-intensity of exercise and inactive exercise mice. Their study indicated a more effective DNA protection for the higher-intensity exercise training. Telomere length can be controlled directly by the telomerase enzyme, htert, and indirectly by a group of proteins called the shelterin complex.
The latter consisted of six proteins: telomere-related factors 1 (TRF1) and 2 (TRF2), TRF1-interacting protein 2 (TIN2), protection of telomeres 1 (Pot-1), the Pot-1- and Tin2-organizing protein repressor/activator protein 1 (RAP1), and tripeptidyl peptidase I (TPP1). To explore the potential of exercise in the maintenance of telomere length, Laye, et al. [29] examined the mRNA and protein levels of proteins associated with the shelterin complex in marathoners. The results showed an increment of mRNA levels of DNA repair enzymes Ku70 and Ku80 and mRNA levels of TRF1, TRF2, and Pot-1 in both skeletal muscle and peripheral blood mononuclear cells in twenty-four hours after marathon exercise. To verify the hypotheses that regulating telomeric gene transcription and/or microRNAs (miRNAs) may be a key molecular mechanism in protection of telomere induced by exercise, Chilton, et al. [30] investigated the acute exercise-induced expression of telomeric genes and miRNAs in volunteers. Blood samples were taken before and after exercise and 60 minutes post-exercise. Results showed the upregulation of telomerase reverse transcriptase (TERT) mRNA and sirtuin-6 (SIRT6) mRNA after exercise. Fifty-six miRNAs were also differentially regulated after exercise. Four miRNAs (miR-186, miR-181, miR-15a and miR-96) were considered as potential targeted telomeric gene mRNA after in silico analysis and all of the four miRNAs exhibited significant upregulation 60 minutes after exercise. Telomeric repeat binding factor 2, interacting protein was identified as a potential binding target for miR-186 and miR-96 and demonstrated concomitant downregulation at the corresponding time point. Their results showed that intense exercise was sufficient to regulate key telomeric genes and miRNAs. Preventing telomere attrition induced by exercise is also considered as a potential molecular mechanism in aged- related diseases treatment.
Exercise Improves Epigenetic Adaptations
Epigenetics is defined as heritable changes in gene function or in the DNA sequence. It suggests that epigenetic alterations possess a heritable characteristic. Mechanisms of epigenetic regulation, including DNA methylation, chromatin remodeling and histone post-translational modifications, and chromatin remodeling (such as miRNA expression changes) can induce heritable phenotypic changes without a change in DNA sequence. Disruption of gene expression patterns which are governed by epigenetics is involved in aging and aged-related diseases including cancer, diabetes, cardiovascular diseases. Although genetic programs undergoing epigenetic changes often occur in aging, aged-related diseases and the aberrations might be pharmaceutically reversed. Intervention- targeting the specific epigenetic mechanisms can be clinically applied in retarding aging and preventing from age-related diseases. Epigenetic regulation is also a pivotal molecular mechanism to explain retardation of aging by exercise.
Utilizing Meta-analysis, Brown, et al. [31] illuminated the effect of exercise on DNA methylation and imprinted genes in skeletal muscle gene networks. The results showed that six imprinted loci (RB1, MEG3, UBE3A, PLAGL1, SGCE, INS) were important for muscle gene networks, while meta-analysis uncovered five exercise-associated imprinted loci (KCNQ1, MEG3, GRB10, L3MBTL1, PLAGL1). Exercise regulates DNA methylation in 60% of imprinted loci. Exercise-associated DNA methylation change was more obvious in older people.
“OMICS” technologies are characterized by high-throughput interfaces and are extensively used in the investigation of genome, epigenome, transcriptome, proteome, and metabolome. Utilizing OMICS techniques, biologists have explored the intricate biological systems and unveil the mystery of the complex cellular phenotypes. New advances in OMICS technologies have offered new opportunities in the detection of genetic, epigenetic and transcriptomic biomarkers. As an important technique of RNomics, small non-coding miRNAs have extensively been utilized. miRNAs regulate gene expression by binding to mRNA to induce degradation or inhibit translation. The researchers have shown that miRNAs affect many processes and play a crucial role in cell differentiation, proliferation, apoptosis, and maintenance of homeostasis. Circulating miRNAs (c-miRNAs) are utilized as biomarkers in physiological conditions such as pregnancy or exercise or aged-related diseases.
The evidence from Denham [32] showed a close relationship between the exercise training and genetic variants and epigenetic modifications including DNA methylation, histone protein modifications, and the regulation of (miRNAs). As non-coding RNAs (ncRNAs), miRNAs regulate gene expression through post- transcriptional mechanisms or orchestrating conformational changes to chromatin. Considering function of miRNAs in intercellular signaling and it is proposed as biomarkers of the health benefits acquired from exercise. Many miRNAs are implicated in various exercise-induced adaptations (e.g., physiological cardiac hypertrophy, mitochondrial biogenesis, insulin sensitivity) [32].
Based on a systemic review on epigenetic mechanism of exercise adaptation in skeletal muscle memory, Sharples, et al. [33] hypothesis states that skeletal muscle is programmable and can ‘remember’ early-life metabolic stimuli including epigenetic information regulating further skeletal muscle function in adult life. The evidence from sports physiological studies showed that muscle can recall the morphological and functional status of an earlier period of exercise when it is stimulated. Epigenetic modification occurs after retraining based on the memory of myonuclei to prior anabolic hormonal/exercise encounters. To explore the effect of exercise on epigenetic adaptation, McGee, et al. [34] estimated global histone modifications, a key molecular event in mediating chromatin remodelling and transcriptional activation and the signalling mechanisms regulating these processes. The results showed that 60 min of acute high-intensity cycling exercise (75% VO2 max/maximal aerobic capacity) induced an increase of lysine 36 which is associated with transcriptional elongation. On the molecular mechanism, they found that exercise induced the export of histone deacetylases (HDAC) 4 and 5 from the nucleus which was involved in removing the transcriptional repressive function. Exercise activated the AMP-activated protein kinase and the calcium-calmodulin- dependent protein kinase II, which induced phosphorylation- dependent class IIa HDAC nuclear export.
Exercise Regulates Nutrient Sensing
Cellular homeostasis including proteostasis is controlled by an evolutionary conserved cellular digestive process called autophagy. This mechanism is tightly regulated by the mechanistic target of rapamycin (mTOR) called as nutrient sensor. mTOR is regard as a pivotal regulator of proteostasis [35]. Exercise is a powerful intervention in regulating nutrient sensing and maintaining proteostasis. mTOR performs a pivotal effect in cellular homeostasis by regulating cellular growth and protein synthesis. It is activated by nutrient (i.e., amino acids) intake, growth factors (i.e., growth hormone [GH], insulin-like growth factor 1 [IGF-1]) stimulation, and mechanical stress, and is inhibited by nutrient deprivation, energetic stress, and rapamycin [36]. Dramatically, mTOR is involved in lifespan and the aging process and inhibition of mTOR pathway is regarded as a potential strategy for retard aging and prolonging lifespan [36]. In another hand, autophagy aroused by mTOR inhibition is also regarded as a pivotal process in prolonging lifespan and preventing aging. Autophagy is involved in nutrient sensing pathway as a key downstream target of the IGF-Akt-mTOR signaling pathway [36].
Caloric Restriction (CR) i.e., Dietary Restriction (DR), a known promoter of lifespan, performs its effect in retarding aging and prolonging lifespan via a mTOR/autophagy nutrient sensing pathway. Exercise and/or CR result in a concomitant downregulation of mTOR and activation of autophagy. It has been increasingly demonstrated that autophagy play a major role in maintaining cellular homeostasis and influencing lifespan and longevity by the degrading misfolded proteins and exhausted organelles. As a hallmark of aging, autophagy is naturally declining over time. A lot of evidence showed that mTOR inhibition and autophagy contributes to molecular mechanisms by which exercise can stop aging and support lifespan extension in rodent and human models. The effect of exercise is similar to those of CR: exerting their effects on autophagy and mTOR through common pathways. AMPK/mTOR signaling may improve insufficient energy metabolism via the autophagy signal pathway. Physical activity can induce autophagy by downregulating the PI3K/Akt/mTOR signaling pathway. Kang, et al. [37] explored the effects of exercise on PI3K/AKT/mTOR signal transmission, autophagy, and cognitive ability in Tg transgenic mice. The results showed that treadmill exercise for 12 weeks 1) improved abnormal mTOR and phosphorylation of PI3K/AKT proteins and inhibited its activity by increased GSK-3β phosphorylation, increased the expression of Beclin-1 protein involved in autophagosome formation and decreased the expression of p62 protein, 3) reversed hyperphosphorylation and aggregation of tau phosphorylated at the site of Ser199/202, Ser404, Thr231, PHF-1 in Tg transgenic mice. Chen et al. [38] showed that chronic treadmill exercise improved learning and memory and potentiated postsynaptic excitation, enhanced neuronal activity of pyramidal neurons, and promoted oligodendrogenesis and axonal myelination at the molecular level. Furthermore, they explored the molecular mechanism. The result showed exercise increased several molecules which are involved in the mTOR pathway including BDNF, which is a potential activator of the mTOR pathway, phosphorylated and total mTOR, phosphorylated and total S6 protein levels, and inhibited elongation factor 4E- BP2. They concluded that exercise improved motor learning via activating the mTOR pathway, which is necessary for spinogenesis, neuronal activation, and axonal myelination.
Exercise Maintains Proteostasis
Proteostasis (i.e., the balance between synthesis and breakdown of proteins) including regulation and control of protein synthesis, conformation, and degradation are needed for ensuring cellular metabolism. The capacity of cells to maintain proteostasis has being gradually declined following aging. Imbalance of proteostasis induces accumulation of damaged proteins and results in aging and aged-related diseases. Anabolic resistance manifests decline of anabolic response to growth signals promoting muscle protein synthesis with aging and explains the phenomenon of age- related muscle loss [39].
Loss of muscle fibre results from anabolic resistance, i.e., imbalances between muscle protein synthesis and breakdown. It is defined as the decline in response to fundamental environmental factors regulating muscle homeostasis (namely physical activity and nutrition), and is also a culprit in catabolic perturbations in muscle proteostasis [40]. Exercise is a powerful intervention in attenuating anabolic resistance 1) produces more energy by promoting catabolism of muscle glycogen and promotes muscle growth by increasing anabolism of protein; 2) keeps proteostasis in muscles stimulated by promoting anabolism of muscles protein in the one hand, and on the another hand, by inhibiting catabolism of muscles protein; 3) promotes dietary amino acid ingestion and enhances post-exercise rates of muscle protein synthesis; and 4) inhibits nutrient receptor, in turn, speeds catabolism of glucose and fatty acid to produce more energy. Generally, anabolism of muscle protein can last for 24- 48 hours after a resistance exercise. Thus, muscle hypertrophy depends on the balance between muscle simulation and nutrient intake in post exercise metabolic processes. Hormones, especially insulin and testosterone and GH are deeply involved in muscle anabolism induced by exercise. Anabolic-Androgenic Steroids (AAS) and other hormones such as GH and IGF-1 have been shown to increase muscle mass as a pharmacological intervention.
Exercise has also been shown to induce significant increase of endogenous hormonal (testosterone (T), GH, IGF-1) elevations.
It is speculated that the hormones increased post-exercise binding to their receptors and upregulate intracellular anabolic pathways [41]. Some biological processes including enhanced protein synthesis, decreased protein breakdown [42], satellite cell activation, Wnt signaling [43], and sex hormone-binding globulin receptor binding [44] contribute to the molecular mechanism of hormonal elevations induced by exercise. The hormones elevated induced by exercise can also promote muscle contraction by increasing intracellular calcium [45]. In addition to steroid hormones, GH and IGF-1 which promote anabolism have also been shown to increase after exercise. McCall, et al.
[46] evaluated concentrations of GH, IGF-1, testosterone, or sex hormone-binding globulin occurred from pre- and post-training and found that acute exercise promotes increase of those hormones and muscle hypertrophy. Mangine, et al. [47] explored the relationships between the endocrine response to resistance exercise and muscle hypertrophy in resistance-trained men, and showed a significant correlation between long-term muscle hypertrophy and elevation of testosterone induced by resistance training.
Atrophy and/or dysfunction of skeletal muscle is one of the aging hallmarks [48]. The size of skeletal muscle mass is regulated by balance between synthesis and degradation of protein. Increased proteolytic pathways (i.e., autophagy and ubiquitin-proteasome system) speed up the loss of muscle mass [49]. Aged population atrophy and/or dysfunction of skeletal muscle is involved in the reduction of mobility and basic activities of daily living. Similarly, age-associated reductions in cardiac systolic performance reduce cardiac output and aerobic exercise capacity. These reductions of skeletal muscles and cardiac muscle with aging are regard as a crime culprit leading to impairments of physiological functions and medical disability.
It is generally believed that skeletal muscle-related properties including grip strength, neuromuscular tasks, and aerobic exercise capacity may be regarded as an independent index predicting survival. Even if a small increase in general muscle function can also indicate a better quality of life and lower risks of mortality [6]. Therefore, there is a clinical need in preventing or delaying the onset of skeletal muscle degeneration in order to extend healthspan. In this regard, exercise has been used as a safe non-pharmacological strategy for attenuating muscle weakness/ wasting in both health and disease. Campos, et al. [50] explored the contribution of exercise in cytosolic protein quality control in a heart failure animal model. The results demonstrated that eight weeks of exercise training can clearly reverse proteasomal insufficiency and relieve exacerbated cardiac oxidative stress and heart failure.
Bozi, et al. [51] found the changes of several biomarkers of muscle protein synthesis, e.g. increasing protein levels of the unfolded protein response markers (GRP78, DERLIN-1 and CHOP), accumulating misfolded and polyubiquitinated proteins, and reducing chymotrypsin-like proteasome activity in myocardial infarction rats. These changes suggested the impairment of cardiac protein quality control. Further study showed that exercise training can reverse these impairments by reducing protein levels of the unfolded protein response markers, and inhibiting accumulation of both misfolded and polyubiquinated proteins, which was associated with restored proteasome activity.
Although enhancement of proteolytic activity has been regarded as a risk leading to skeletal muscle atrophy, autophagy, which disposes cytotoxic proteins and non-functioning organelles, is also needed for the maintenance of muscle proteostasis. One of the effects produced by exercise is that exercise activates autophagy and re-establishes proteostasis [52]. Campose, et al [11] explored the significance of exercise between autophagy and proteostasis in skeletal muscle utilizing a model of neurogenic myopathy in rats. Neurogenic myopathy is a progressive atrophy and impaired contractility disease characterized by accumulation of autophagy-related markers and elevation of damaged proteins, chaperones and pro-apoptotic markers. Their study showed that exercise training prior to neurogenic myopathy can protect from skeletal muscle autophagic flux and loss of proteostasis and accompanied with attenuation of muscle atrophy and better contractility properties.
In a randomized, controlled trial, Witrard, et al. [53] explored the dose-response relationship between whey protein and postabsorptive rates of myofibrillar protein synthesis after exercise and showed that 20-g whey protein was enough for maximal postabsorptive rates of myofibrillar protein synthesis. Similarly, based on re-analysis of the published literature, Moore [54] suggested that a single meal intake of 0.31g/kg of high quality protein should be considered as a nutritional guideline aiming to maximize post-exercise myofibrillar protein synthesis while minimizing irreversible amino acid oxidative catabolism.
Hormones promote protein biosynthesis in muscle via various molecular pathways 1) activating adult stem cells (ASCs); 2) triggering genomic and non-genomic signaling; 3) interacting with other anabolic signaling pathways; and 4) upregulating or downregulating the androgen receptor [55]. Three hormones including testosterone, GH, and IGF are deeply involved in the anabolism induced by exercise. Zeng, et al. [56] investigated the effects of exogenous androgen and resistance exercise on skeletal muscle hypertrophy and the role of mTOR signalling during the process. They found hypertrophy of skeletal muscle and a higher myosin heavy chain expression induced by exercise and dihydrotestosterone implantation. The mTOR molecular pathway was involved in the anabolism of skeletal muscle. Although dihydrotestosterone and exercise can severely increase the mRNA expression of androgen receptor and IGF-I and three peptides related to mTOR signaling including the mTOR phosphorylated, p70S6K, and 4EBP1, combination of dihydrotestosterone and exercise can increase the expression of the mRNA of those molecules more.
Exercise enhances androgen-induced rapid anabolic action, which involves activation of the mTOR pathway. To evaluate effects of resistance exercise training on circulating anabolic hormones (e.g., GH, IGF-1, free IGF-1, testosterone, free testosterone, dehydroepiandrosterone, dihydrotestosterone, and luteinizing hormone), Morton, et al. [57] compared those hormones in circulation and muscle, and intramuscular hormone- related variables in men who had 12 weeks of resistance exercise training. The results indicate that intramuscular androgen receptor content, but neither circulating nor intramuscular hormones (or the enzymes regulating their intramuscular production), contributed to skeletal muscle hypertrophy induced by resistance exercise.
Exercise Enhances Mitochondrial Function
Mitochondria play an essential role in energy metabolism and are also involved in the extensive cellular processes including the oxidation of fatty acids, the metabolism of amino acids, the control of apoptosis, the synthesis of steroid hormone, and the regulation and control of hormonal signaling. Mitochondria maintains its own 16.5-kb genome for the expression of mitochondrial proteins [58]. The sophisticated coordination of both mtDNA and nDNA genomes is necessary to regulate organelle abundance and involved in mitochondrial health, disease and turnover. Mitochondrial health is a key cellular mediator across a range of tissues, and contributes to whole- body vitality. The mitochondrial dysfunction is regarded as a risk molecular event for aging and age-related diseases [59]. On the conditions that perturb metabolic homeostasis, such as exercise or aging, mitochondrial homeostasis is rebalanced by increase in synthesis and fusion, or fission and mitophagy, respectively [60]. Potential molecular mechanism of mitochondrial aging includes the accumulation of DNA mutations induced by decline in electron transport function and the imbalance of quality control mechanisms (e.g. mitophagy and proteolysis) induced by damage of the mitochondrial unfolded protein response [61].
Considering the possible relationship between rejuvenation of mitochondria and enhancement of cellular activity the association between mitochondria and the aging process has been extensively investigated. Mitochondrial quality control might be an effective strategy to combat aging. Pharmacologic activation of mitophagy is another approach that might be widely beneficial in preventing against age-related neurodegenerative disorders and retarding aging [62]. A remarkable characteristic of the mitochondrion is its plasticity, i.e., adjustments of its volume, structure and capacity based on circumstances such as exercise. Maintenance of mitochondrial functions depends on balance between dynamic processes of biogenesis and fusion, and opposing processes of fission and mitophagy. Exercise can conveniently promote robust mitochondrial adaptations. Hence, exercise remains the most potent therapeutic strategy for the improvement of mitochondrial health, not only in muscle, but potentially also in other tissues [63]. Exercise is benefit for maintaining mitochondrial health by inducing obvious changes of mitochondria in number and size. Possible molecular mechanisms include 1) enhancing Ca2+ release from the sarcoplasmic reticulum and enhancing muscle force, as well as the generation of ATP from respiring mitochondria; 2) activating the signalling kinases and increasing the expression of mtDNA transcription factors; 3) activating the UPRmt and further upregulating transcription of mitochondrial chaperones and proteases and equipping the organelle with an augmented capacity for protein folding; 4) enhancing expression of mitochondrial fusion proteins Mfn1/2 along with Opa1 and further facilitating the fusion of the outer and inner membranes, respectively, and improving of metabolites of neighbouring organelles; 5) enhancing mitophagy to clean out the damaged organelles and activating lysosomes to degrade the dysfunctional mitochondria and release the constituent amino acids for cellular recycling; and 6) increasing PGC-1α signalling for mitochondrial biogenesis and fission. Robinson, et al. [64] assessed the effects of exercise including high-intensity aerobic interval, resistance, and combined exercise training on skeletal muscle adaptations in young and older adults.
The results showed that training or exercise for 12 weeks enhanced insulin sensitivity and muscles mass. High-intensity aerobic training and combined training or resistance training improved aerobic capacity and skeletal muscle mitochondrial respiration. High-intensity aerobic exercise revealed a more robust increase in gene transcripts than other exercise modalities. Several age-related changes in the proteome, and mitochondrial functions were in accompanied with increased mitochondrial protein synthesis after high-intensity aerobic training. In conclusion, high-intensity exercise can effectively prevent aged-related decline in muscle mitochondria. Campos, et al. [65] explored the effect of exercise to protect against cardiovascular disease via cardiac autophagy and its contribution to mitochondrial quality control, bioenergetics and oxidative damage in a post-myocardial infarction-induced heart failure in rats. The study showed a reduced mitophagy flux, i.e., accumulation of autophagy-related markers induced by myocardial infarction. The study further showed an accumulation of fragmented mitochondria accompanied by reduction of O2 consumption, elevation of H2O2 release and increase of Ca2+- induced mitochondrial permeability transition pore opening.
The study validated the hypothesis that disruption of mitophagy flux was sufficient to damage cardiac mitochondrial function and increase cardiomyocyte toxicity induced by mitochondrial stress. Further study showed effect of exercise to protect against mitochondrial damages induced by myocardial infarction. Impairment of cardiac mitophagic flux occurred in myocardial infarction 4 weeks rats can be reversed via 8 weeks exercise training. Exercise training also reduced mitochondrial number:size ratio, increased mitochondrial bioenergetics and better cardiac function. Interestingly, exercise training increased cardiac mitochondrial number, size and oxidative capacity, but did not affect mitophagic flux in sham-treated animals. In conclusion, the potential contribution of exercise in restoring cardiac mitophagy flux in heart failure is associated with better mitochondrial quality control, bioenergetics and cardiac function.
As an oxidative stress-sensitive gene localized to mitochondria, CDGSH iron sulfur domain 2 (Cisd2 ) not only regulates mitochondrial activity and biogenesis, but also contributes to extension of the lifespan. [66]. A lack of Cisd2 shortened lifespan in mice and Cisd2 transgenic mice lived longer and healthier [67]. Cisd2 plays a key role in sustaining metabolic activity, ameliorating aging-associated mitochondrial dysregulation, reducing DNA damages and improving the calcium imbalance [66]. Yokokawa, et al. [68] explored the effect chronic exercise training on expression of Cisds and mitochondrial proteins. The results showed that chronic exercise increased Cisd 1 and 2 as well as mitochondrial protein expression. Further, they showed that mitochondrial biogenesis induced by chronic exercise coincides with the expression of Cisds.
Exercise Protects Against Cellular Senescence
Cellular senescence, a trigger of growth arrest of cells, contributes to physiological dysfunction with aging and promotes age-related pathologies [69]. Cellular stressors including DNA damage, dysfunctional telomeres, oxidative stress, or metabolic stimuli to induce cell cycle arrest and the cellular signaling cascades to activate p53/p21 and p16 tumor suppressor are regarded as a common pathway leading to cellular senescence [70]. The proinflammatory and prooxidative Senescence- Associated Secretory Phenotype (SASP) impair neighboring cells and boost cellular senescence [71].
Therapy strategies delaying cellular senescence should include pro-senescent therapies and antisenescent therapies. The former prevents from senescence by limiting proliferation and fibrosis. The latter prevents from senescence by eliminating accumulated senescent cells and recovering tissue function [70]. Exercise which prevents and reduces indicators of cellular senescence is an irrefutable candidate for therapy strategy of cellular senescence. Schafer et al. [72] explore effects of exercise preventing premature senescent cell accumulation and systemic metabolic dysfunction induced by a Fast-Food Diet (FFD). Their study showed that FFD causes harmful changes in physical and metabolic functions which were associated with dramatic increases in several markers of senescence, including p16, Enhanced Green Fluorescent Protein (EGFP), senescence- associated β-galactosidase, and SASP in transgenic mice that express EGFP in response to activation of the senescence- associated p16INK4a promoter.
Further, Schafer, et al. showed that exercise prevents against the accumulation of senescent cells and the expression of p16, EGFP, senescence-associated β-galactosidase, and the SASP accompanied by attenuating the effects of the FFD on health. To verify the hypothesis that exercise prevent from senescence and dysfunction of vascular endothelial cells, Rosseman, et al. [73] tested comparatively the expression of proteins related to cellular senescence in adults and older sedentary and in adults and older habitually exercising. The results showed an increasing protein expression of p53, a transcription factor related to increased cellular senescence, and the Cyclin-Dependent Kinase Inhibitors (CDKI) p21 and p16 in Endothelial Cells (ECs) obtained from antecubital veins of older sedentary (60 ± 1 yr) as compared to young sedentary (22 ± 1 yr) adults. There are no increases of these age-related biomarkers in older exercising adults (57± 1 yr) as compared to young sedentary. The protein levels of p53, p21, and p16 were inversely associated with vascular endothelial function. Similarly, protein expression of p53 and p21 was 26% and 23% higher, respectively, in ECs sampled from brachial arteries of healthy older sedentary (63 ± 1 yr) versus young sedentary (25 ± 1 yr) adults; age-related changes in arterial EC p53 and p21 expression were not observed in older habitually exercising adults (59 ± 1 yr). The study indicated that EC senescence is associated with sedentary aging and is linked to endothelial dysfunction. Moreover, these data suggest that aerobic exercise prevent EC senescence by protecting against endothelial dysfunction with age. Philippe, et al. [74] investigated the effect of exercise training on the T-cell senescence in pre- diabetic subjects. The results showed an increase of the CD4+/ CD8+ and CD4+/CD3+ ratio and a decrease of the CD8+/CD3+ ratio after exercise training.
The results showed a proportional increase of CD4+/ CCR7+/CD45RO+ central memory cells, naive CD8+/CCR7+/ CD45RO- and CD8+/CCR7+/CD45RO+ central memory cells, while proportions of CD4+/CCR7-/CD45RO- TEMRA cells (-2.17 ± 4.66%; p = 0.012), CD8+/CCR7-/CD45RO- TEMRA cells and CD16+ cells decreased after training. All of those changes suggested that exercise training stimulates production and mobilization of naive T-cells and reverses hallmarks of T-cell senescence in pre-diabetic subjects. To verify the hypothesis that neuroprotective effect of endurance exercise is associated with cellular senescence, neuroinflammation, and oxidative stress, Jang et al. [75] tested markers of cellular senescence (p53, p21, p16, β-galactosidase, and lipofuscin), markers of neuroinflammation including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and markers of oxidative stress (TNF receptor-associated factor 6, c-Jun N-terminal kinase, and c-Jun). The results showed that endurance exercise ameliorated weight gain, fasting blood glucose levels, and visceral fat gain in a mouse model of obesity induced by a high-fat+high-fructose diet. At molecular level, they showed that exercise reduced the expression of markers of cellular senescence: p53, p21, p16, β-galactosidase, and lipofuscin in the hippocampus. Further study showed that endurance exercise prevented high-fat+high- fructose diet-induced neuroinflammation (e.g., TNF-α and IL-1β) by inhibiting toll-like receptor 2 downstream signaling cascades (e.g., TNF receptor-associated factor 6, c-Jun N-terminal kinase, and c-Jun) in parallel with reduced reactive glial cells. This anti- inflammatory effect of endurance exercise was associated with the reversion of high-fat+high-fructose diet-induced cellular oxidative stress.
Exercise Reduces SCs Exhaustion
ASCs (progenitor cells) are a cluster of cells located in tissue- specific niches and keep a potential ability to differentiate to all kinds of cell types [76]. ASCs including satellite cells, hematopoietic SCs, skin SCs, intestinal SCs, and germline SCs contribute to the regeneration, homeostasis, and aging of various tissues [77]. An obvious characteristic of aging is gradual decline of tissues and organs functions with aging. ASCs are responsible for the repair and the regeneration of cells and the maintenance of tissue and organs. Unfortunately, the amount and potential regenerative ability of ASCs are gradually being lost with aging. As a result, organs gradually degrade and living organisms die eventually. Regenerative function of ASCs declines with aging and the decline of ASCs promotes organ senescence [78].
ASCs exhaustion is characterized by the progressive loss of physiological functions and the accumulation of the impairment of tissue and organ function that results in the development of multiple pathologies and age-related diseases and is regarded as a pivotal hallmark of aging [79]. To verify the hypothesis, Vilas, et al. [79] explored the relationship between accelerated SCs depletion and premature aging in a mouse model in which adult Sox2+SCs depletion was induced. They found the relationship between signs of premature aging (e.g. increased kyphosis and hair graying, and reduced fat mass) and a short period of partial repetitive depletion of ASCs and the relationship between cellular senescence and partial repetitive Sox2+cell depletion. Further, they prolonged a protocol of partial repetitive depletion of Sox2+cells to force regeneration from the remaining Sox2+cells and to induce their exhaustion. They verified that ASCs exhaustion can lead to cellular senescence induction and premature aging based on senescence specific staining and the analysis of the expression of genetic markers. They study demonstrated that ASCs depletion trigger senescence induction and premature aging of tissues, and physiological aging.
Both of acute exercise bout and regular exercise training promote proliferation of ASCs and increase circulating haematopoietic stem and progenitor cells in athletes, healthy individuals, and in individuals suffering from aged-related diseases [80]. The investigations have showed that individuals who undergo regular and systematical exercise training have a higher level of circulating haematopoietic stem and progenitor cells than sedentary individuals [81]. To explore the effects of exercise on aged SCs function in the context of tissue regeneration, Brett, et al [82] investigated comparably muscle SCs function and muscle regeneration in young adult and old mice with and without rotating running wheels. The results showed that exercise in the form of voluntary wheel running accelerates muscle repair in old animals and improves old muscle SCs function. Utilizing transcriptional profiling and genetic analyses, they furthermore verified the restoration of cyclin D1, a biomarker for decline of muscle SCs with aging, as a key molecular event for rejuvenation of old muscle SCs activation ability.
ASCs play a major role in maintenance of tissues or cells, reconstruction and regeneration of multiple tissues and especially in replenishment of dead cells from apoptosis. In a recent paper, Sen et al. [83] elaborated the relationship between rejuvenation of ASCs including endothelial progenitor cells and SMCs and aerobic exercise. Metabolic disorders like diabetes dramatically decrease endothelial progenitor cell numbers and impair its function. Increasing endothelial progenitor cell number and function has the potential to be a treatment strategy for both diabetes and cardiovascular disease. Exercise can effectively improve the production and increases numbers of endothelial progenitor cells. At molecular level, Nitric Oxide (NO) contributes to production and development of endothelial progenitor cells, which may be secondary to its antiapoptotic effect [84]. Similarly, exercise helps to reduce phosphoinositide 3-kinase mediated apoptosis which depends on NO [85].
Prostaglandin E1 (PGE1), Vascular Endothelial Growth Factor (VEGF) and, and Stromal Derived Factor 1 (SDF 1) are considered as the biomarkers of ASCs based on its bioactivity in mediating upregulation of endothelial progenitor cells [86]. Exercise increases PGE1, VEGF, and SDF1 levels [86]. Schlager, et al. [87] explored effects of exercise preventing atherosclerosis based on ASCs development. The results showed that exercise promoted angiogenesis and improved endothelial function and decreased atherosclerosis. At molecular level, exercise mobilized endothelial progenitor cells and reduces asymmetric dimethylarginine an endogenous inhibitor of NO synthase. Mobilization of ASCs stimulated by exercise depended on exercise intensity. Laufs, et al. [88] verified that circulating endothelial progenitor cell numbers increased after intensive and moderate exercise activity for 30 minutes, but it did not change after the exercise in less than 10 minutes. By comparing effect of aerobic-endurance exercise in middle-aged and older healthy men, Hoetzer, et al. [89] concluded that regular aerobic-endurance exercise is a reliable treatment strategy in retarding aging by increasing endothelial progenitor cells clonogenic and migratory capacity.
Mesenchymal stromal cells (MSCs) are multipotent cells which can differentiate into adipocytes, chondrocytes, and osteoblasts and are defined by specific markers e.g. CD90, CD44, CD105, and CD73, but not CD45, CD31, and CD34 [90]. Studies showed that exercise may facilitate MSCs migration by increasing IL-6 and recruit SCs [91]. It was reported that MSC secretome is responsible for hematopoietic stem and progenitor cells (HSPC) mobilization and proliferation and exercise induce homing of HSPCs to extramedullary sites [92]. Studies showed that exercise increased therapeutic MSC transplantation in traumatic brain injury [93] and ischemic cerebral cortex [94] in the rat model. Exercise plays a vital role in differentiation of multipotent MSCs in bone. Li, et al. [95] examined the effects of mechanical strain on osteogenic and adipogenic differentiation of cultured MSCs using a mechanical strain device. Runt-related transcription factor2 (Runx2), Osterix (Osx), and I-collagen were tested as osteogenic differentiation markers and peroxisome proliferator-activated receptor (PPAR)γ-2, CCAAT/enhancer binding protein (C/EBP) α, and lipid droplets were tested as adipogenic differentiation markers. The results showed that the expression of Runx2, Osx, and I-collagen protein was induced by strain stimulation. Oppositely, the expression of PPARγ-2 and C/EBPα was inhibited by strain stimulation. Their study verified that exercise can promote differentiation of MSCs into osteoblasts and can impede differentiation into adipocytes. The adult human heart possesses a limited regenerative potential. The activation of cardiac- resident SCs ensures cardiac cells regeneration and hypertrophy [96]. Regeneration and growth of skeletal muscle are mainly controlled by resident SCs (e.g. satellite cells) [97]. Exercise induces muscle hypertrophy by activating satellite cells in mature skeletal muscle cells and therefrom promoting differentiation of muscle cells [98]. On molecular levels, the expression of VEGF, IGF, NO and hepatocyte GF, fibroblast GF, which are endocrine and locally expressed autocrine and paracrine GF, contribute to an activation of the satellite cells in exercise status [97].
Macaluso, et al. [99] reviewed the contribution of exercise to the regeneration of ASCs in the various tissue including heart, bone, liver, brain, and muscle etc. They concluded that the same factors that stimulate a high proliferation of SCs in skeletal muscle after exercise should also stimulate the proliferation of ASCs in the tissue in which they reside, such as heart, bone, liver and brain etc. Regular exercise retards aging and prevents aged-related diseases by maintaining a higher ASCs reserve and progenitor cell potential for rapid activation in response to stress and damage. O’Reilly, et al. [100] explored effects of muscles lengthening contractions on satellite cell activation and the expression of 8 related proteins including hepatocyte growth factor, hepatocyte growth factor activator, hepatocyte growth factor activator inhibitor-1, and hepatocyte growth factor activator inhibitor-2 in both of skeletal muscle, and serum. The results showed an increase of SCs [neural cell adhesion molecule (NCAM)-labeled cells] at 24 hours and a peak value at 72 hours after 300 lengthening contractions. The expression of 8 related proteins is in line with an increase of ACs after muscle contractive training. Hence, a single bout of lengthening muscle contractions is enough to activate SCs via both a local and systemic hepatocyte growth factor response. In a recent study, Frodermann, et al. [101] showed that exercise protected from atherosclerosis by chronic leukocytosis but emergency hematopoiesis in mice. At molecular mechanism, they showed that exercise diminished leptin production in adipose tissue, augmented quiescence- promoting hematopoietic niche factors in leptin-receptor- positive stromal bone marrow cells.
Some growth factors induced by exercise have effects on SCs proliferation. Both VEGF and IGF-1 plays a key role in promoting angiogenesis and hippocampal neurogenesis based on their effect on ASCs proliferation [102]. Exercise can increase VEGF expression, in turn, trigger release of haematopoietic and endothelial progenitor cells from the bone marrow [103]. Utilizing VEGF gene-ablated mice, Rich, et al. [104] explored the effect of VEGF produced by skeletal myofibers in regulating hippocampal neuronal precursor cell proliferation following exercise training. The results suggested that VEGF originated from skeletal myofibers is abundantly released during exercise and promotes ASCs proliferation, and results in angiogenesis and hippocampal neurogenesis. IGF-1 regulates skeletal muscle growth by triggering the PI3K/Akt signaling cascade to promote protein synthesis. Resistance exercise increases IGF- 1 concentration in the blood stream, skeletal muscle, and brain [105].
The growth factor BDNF contributes to neuroplasticity, which describes a responsive ability of the brain to environment changes, e.g. exercise. A recent study from Quan [106] showed that exercise promotes proliferation and differentiation of neuronal SCs induced by BDNF. Leukemia Inhibitory Factor (LIF) has a range of biological roles including platelet formation, proliferation of haematopoietic cells, bone formation, neural survival and formation, and muscle and satellite cell proliferation. A study from Broholm, et al. [107] showed that exercise promoted satellite cell proliferation which was mediated by LIF. Platelet- Derived Growth Factor (PDGF) is also a factor to promote the proliferation and migration of MSCs. Farup, et al. [108] showed that PDGF was involved in the effects of resistance training on proliferation of mesenchymal progenitor cell proliferation, and expansion of ASCs pool.
Exercise Improves Intercellular Communication
Intercellular communication is a basic biological process by which various messages are transferred between cells. The soluble factors are one of the best characterized styles of intercellular communication which perform their effect on the neighboring cells [109]. Senescence is essentially a stable cell cycle arrest induced by the cell cycle inhibitors p21CIP, p53, and p16INK4A [110]. Senescent cells arrest not only cell cycles themselves, but also trigger senescence of bystander cells through intercellular communication based on the secretion of bioactive molecules, e.g. SASP factors [111]. Many signalling factors including pro- inflammatory chemokines, interleukins, and cytokines, which are regarded as SASP factors, are involved in the senescence process [112]. SASP factors are regard as the ‘soul’ of senescence. The classic SASP factors include soluble factors, growth factors, and extracellular matrix remodeling enzymes [113].
Recent studies pay more attention to extracellular vesicles (exosomes), which are also involved in senescence-associated process via intercellular communication style. Exosomes are nano-sized vesicles that serve as mediators for cell-to-cell communication. Exosomes are released during exercise as a key mediator to be involved in communication between cells. Owing to loading of proteins, nucleic acids, and lipids compositions, exosomes are characterized as a profile of producer cells. Hence, they can be utilized as a potential cell-free therapeutic strategy [114]. Based on originating cells status, exosomes alleviating senescence cells by transferring the phenotypes related to senescence. Hence, exosomes are also regarded as age-related disease markers [115]. Age-related changes of exosomes include1) increase of release during senescence of fibroblasts, epithelial cells, and cancer cells [116]; 2) increase with aging [117]; and change of composition (miRNAs, proteins, or lipids) with aging [118]. Among exosomes derived from various cellular origins, MSC-derived exosomes (MSC-exosomes) have been focused based on their immunomodulatory and regenerative characteristics. Studies have shown effects of MSC-exosomes in anti-inflammatory, anti-aging and wound healing in vitro and in vivo models [112]. A recent study from Willis, et al. [119] showed that MSC-exosomes can revise lung injuries: attenuate alveolar damage, improve pulmonary fibrosis, remodel capillary and enhance exercise ability in mice. Utilizing meta analysis, Estébanez, et al. [120] systematically surveyed the effects of exercise on exosome release. The results showed that exercise promotes the release of exosomes without modification of its vesicle size. Further analysis on molecular mechanism showed that exercise promoted release of exosome markers (CD63, Alix, CD81, and Flot-1) along with other exosome-carried proteins into circulation. Exercise promoted proliferation and differentiation of skeletal muscle by increasing release of exosome-carried miRNAs, including miR-1, miR-133a, miR-133b, miR-206, and miR-486.
Exercise Improves Decline of Organs and Molecular Hallmarks of Aging
Exercise can effectively prevent from decline of organs and molecular hallmarks of secondary aging. Aging is characterized by decline of organs and systems: brain atrophy, neurofibrillary tangles and senile plaques in brain; decrease of maximal breathing capacity and maximum cardiac output in lungs and heart; accumulation of fat and decline of hepatotoxin elimination in liver, insulin resistance and weakening of digestion and absorption; weakening of poison elimination in kidneys; neurodegeneration and abnormal neuroregulation; and decrease of hormones related to growth and development and metabolism. All of those declines with aging can be alleviated by exercise. At molecular level secondary aging are characterized by nine cellular and molecular hallmarks: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cells exhaustion, and altered intercellular communication. All of nine cellular and molecular hallmarks can be attenuated by exercise.
CONCLUSION
Although we can’t arrest primary aging by revising genetic program, it is believed that we can attenuate pathophysiological events of secondary aging to prolong lifespan and improve life quality by preventing all of nine hallmarks of aging. Exercise is a valid intervention without any side effects for life extension and life quality improvement. At molecular level exercise can arrest all of nine hallmarks of aging: maintaining genomic stability; attenuating telomere attrition; preventing epigenetic alterations; maintaining proteostasis; regulating nutrient sensing; enhancing the mitochondrial function; delaying cellular senescence; reducing SCs exhaustion; altering intercellular communication. It is hypothesized that improvement of nine molecular and cellular hallmarks of aging which is improved by exercise is to be consistent with higher metabolic level in exercise. To keep a higher metabolic level, every organ and system has to enhance its turnover. It means that every organ and system of an older person has to keep a metabolic work load similar to those of a youngster. Hence cellular and molecular events happened in exercise tend to a younger status.
Exercise can miraculously prevent against all nine hallmarks of aging at molecular level. It indicates a possibility that almost all age-related diseases can be prevented by exercise. It also encourages us to develop a strategy how exercise projects (exercise intensity, style and rhythm) can effectively perform its protective effect for aging and aged-related diseases.
REFERENCES
- Garatachea N, Lucia A. Genes, exercise and ageing. Ageing Res Rev. 2013; 12(1): 90-102. doi: 10.1016/j.arr.2012.09.003.
- Azzolino D, Arosio B, Marzetti E, Calvani R, Cesari M. Nutritional Status as a Mediator of Fatigue and Its Underlying Mechanisms in Older People. Nutrients. 2020; 12(2): 444. doi: 10.3390/nu12020444. PMID: 32050677; PMCID: PMC7071235.
- Hilfiker R, Meichtry A, Eicher M, Nilsson Balfe L, Knols RH, Verra ML, et al. Exercise and other non-pharmaceutical interventions for cancer-related fatigue in patients during or after cancer treatment: a systematic review incorporating an indirect-comparisons meta- analysis. Br J Sports Med. 2018; 52(10): 651-658. doi: 10.1136/ bjsports-2016-096422. Epub 2017 May 13. PMID: 28501804; PMCID: PMC5931245.
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153(6): 1194-1217. doi: 10.1016/j. cell.2013.05.039. PMID: 23746838; PMCID: PMC3836174.
- Pavanello S, Angelici L, Hoxha M, Cantone L, Campisi M, Tirelli AS, et al. Sterol 27-Hydroxylase Polymorphism Significantly Associates With Shorter Telomere, Higher Cardiovascular and Type-2 Diabetes Risk in Obese Subjects. Front Endocrinol (Lausanne). 2018; 9: 309. doi: 10.3389/fendo.2018.00309. PMID: 29951035; PMCID: PMC6008574.
- Rebelo-Marques A, De Sousa Lages A, Andrade R, Ribeiro CF, Mota- Pinto A, Carrilho F, et al. Aging Hallmarks: The Benefits of Physical Exercise. Front Endocrinol (Lausanne). 2018; 9: 258. doi: 10.3389/ fendo.2018.00258. PMID: 29887832; PMCID: PMC5980968.
- Seals DR, Justice JN, LaRocca TJ. Physiological geroscience: targeting function to increase healthspan and achieve optimal longevity. J Physiol. 2016; 594(8): 2001-24. doi: 10.1113/jphysiol.2014.282665. Epub 2015 Mar 11. PMID: 25639909; PMCID: PMC4933122.
- Gomez-Cabrera MC, Domenech E, Viña J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med. 2008; 44(2): 126-131. doi: 10.1016/j. freeradbiomed.2007.02.001. Epub 2007 Feb 9. PMID: 18191748.
- Arsenis NC, You T, Ogawa EF, Tinsley GM, Zuo L. Physical activity and telomere length: Impact of aging and potential mechanisms of action. Oncotarget. 2017; 8(27): 45008-45019. doi: 10.18632/ oncotarget.16726. PMID: 28410238; PMCID: PMC5546536.
- Ling C, Rönn T. Epigenetic adaptation to regular exercise in humans. Drug Discov Today. 2014; 19(7): 1015-1018. doi: 10.1016/j. drudis.2014.03.006. Epub 2014 Mar 14. PMID: 24632002.
- Campos JC, Baehr LM, Gomes KMS, Bechara LRG, Voltarelli VA, Bozi LHM, et al. Exercise prevents impaired autophagy and proteostasis in a model of neurogenic myopathy. Sci Rep. 2018; 8(1): 11818. doi: 10.1038/s41598-018-30365-1.
- Coiro V, Volpi R, Gramellini D, Maffei ML, Volta E, Melani A, et al. Effect of physical training on age-related reduction of GH secretion during exercise in normally cycling women. Maturitas. 2010 Apr;65(4):392-5. doi: 10.1016/j.maturitas.2009.12.020. Epub 2010 Feb 1. PMID: 20117890.
- Hood DA, Memme JM, Oliveira AN, Triolo M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu Rev Physiol. 2019; 81: 19-41. doi: 10.1146/annurev-physiol-020518-114310.Epub 2018 Sep 14. PMID: 30216742.
- Song Z, von Figura G, Liu Y, Kraus JM, Torrice C, Dillon P, et al. Lifestyle impacts on the aging-associated expression of biomarkers of DNA damage and telomere dysfunction in human blood. Aging Cell. 2010; 9(4): 607-615. doi: 10.1111/j.1474-9726.2010.00583.x. Epub 2010 Jun 17. PMID: 20560902; PMCID: PMC2910221.
- Shefer G, Rauner G, Stuelsatz P, Benayahu D, Yablonka-Reuveni Z. Moderate-intensity treadmill running promotes expansion of the satellite cell pool in young and old mice. FEBS J. 2013; 280(17): 4063- 4073. doi: 10.1111/febs.12228. Epub 2013 Apr 12. PMID: 23464362; PMCID: PMC3711960.
- Ball D. Metabolic and endocrine response to exercise: sympathoadrenal integration with skeletal muscle. J Endocrinol. 2015; 224(2): R79-R95. doi: 10.1530/JOE-14-0408. Epub 2014 Nov27. PMID: 25431226.
- Vilela TC, Muller AP, Damiani AP, Macan TP, da Silva S, Canteiro PB, et al. Strength and Aerobic Exercises Improve Spatial Memory in Aging Rats Through Stimulating Distinct Neuroplasticity Mechanisms. Mol Neurobiol. 2017; 54(10): 7928-7937. doi: 10.1007/s12035-016- 0272-x. Epub 2016 Nov 22. PMID: 27878552.
- Zhu D, Tian J, Wu X, Li M, Tang X, Rui K, et al. G-MDSC-derived exosomes attenuate collagen-induced arthritis by impairing Th1 and Th17 cell responses. Biochim Biophys Acta Mol Basis Dis. 2019; 1865(12): 165540. doi: 10.1016/j.bbadis.2019.165540. Epub 2019 Aug 27. PMID: 31470074.
- Gasparovic AC, Milkovic L, Sunjic SB, Zarkovic N. Cancer growth regulation by 4-hydroxynonenal. Free Radic Biol Med. 2017; 111: 226-234. doi: 10.1016/j.freeradbiomed.2017.01.030. Epub 2017 Jan 25. PMID: 28131901.
- Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med. 2014; 16(1): 161-174. doi: 10.1007/s12017-013-8270-x. Epub 2013 Oct 10. PMID: 24114393; PMCID: PMC3948322.
- Marais L, Stein DJ, Daniels WM. Exercise increases BDNF levels in the striatum and decreases depressive-like behavior in chronically stressed rats. Metab Brain Dis. 2009; 24(4): 587-597. doi: 10.1007/ s11011-009-9157-2. Epub 2009 Oct 21. PMID: 19844781.
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, ET AL. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011; 108(10): 4135-4140. doi: 10.1073/ pnas.1019581108. Epub 2011 Feb 22. PMID: 21368114; PMCID: PMC3053975.
- Radák Z, Naito H, Kaneko T, Tahara S, Nakamoto H, Takahashi R, ET AL. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflugers Arch. 2002; 445(2): 273-278. doi: 10.1007/ s00424-002-0918-6. Epub 2002 Sep 13. PMID: 12457248.
- Dimauro I, Sgura A, Pittaluga M, Magi F, Fantini C, Mancinelli R, ET AL. Regular exercise participation improves genomic stability in diabetic patients: an exploratory study to analyse telomere length and DNA damage. Sci Rep. 2017; 7(1): 4137. doi: 10.1038/s41598-017-04448-4. PMID: 28646223; PMCID: PMC5482873.
- Denham J, O’Brien BJ, Charchar FJ. Telomere Length Maintenance and Cardio-Metabolic Disease Prevention Through Exercise Training. Sports Med. 2016; 46(9): 1213-1237. doi: 10.1007/s40279-016-0482-4. PMID: 26914269.
- Muniesa CA, Verde Z, Diaz-Ureña G, Santiago C, Gutiérrez F, Díaz E, et al. Telomere Length in Elite Athletes. Int J Sports Physiol Perform.2017; 12(7): 994-996. doi: 10.1123/ijspp.2016-0471. Epub 2016 Dec5. PMID: 27918657.
- Simoes HG, Sousa CV, Dos Santos Rosa T, da Silva Aguiar S, Deus LA, Rosa ECCC, et al. Longer Telomere Length in Elite Master Sprinters: Relationship to Performance and Body Composition. Int J Sports Med. 2017; 38(14): 1111-1116. doi: 10.1055/s-0043-120345. Epub 2017 Nov 3. PMID: 29100249.
- de Carvalho Cunha VN, Dos Santos Rosa T, Sales MM, Sousa CV, da Silva Aguiar S, et al. Training Performed Above Lactate Threshold Decreases p53 and Shelterin Expression in Mice. Int J Sports Med. 2018; 39(9): 704-711. doi: 10.1055/a-0631-3441. Epub 2018 Jun 26. PMID: 29945271.
- Laye MJ, Solomon TP, Karstoft K, Pedersen KK, Nielsen SD, Pedersen BK. Increased shelterin mRNA expression in peripheral blood mononuclear cells and skeletal muscle following an ultra-long- distance running event. J Appl Physiol (1985). 2012; 112(5): 773- 781. doi: 10.1152/japplphysiol.00997.2011. Epub 2011 Dec 8. PMID: 22162529.
- Chilton WL, Marques FZ, West J, Kannourakis G, Berzins SP, O’Brien BJ, et al. Acute exercise leads to regulation of telomere-associated genes and microRNA expression in immune cells. PLoS One. 2014; 9(4): e92088. doi: 10.1371/journal.pone.0092088. PMID: 24752326; PMCID: PMC3994003.
- Brown WM. Exercise-associated DNA methylation change in skeletal muscle and the importance of imprinted genes: a bioinformatics meta-analysis. Br J Sports Med. 2015; 49(24): 1567-1578. doi: 10.1136/bjsports-2014-094073.
- Denham J, Gray A, Scott-Hamilton J, Hagstrom AD. Sprint Interval Training Decreases Circulating MicroRNAs Important for Muscle Development. Int J Sports Med. 2018; 39(1): 67-72. doi: 10.1055/s- 0043-120763. Epub 2017 Nov 10. PMID: 29126336.
- Sharples AP, Stewart CE, Seaborne RA. Does skeletal muscle have an ‘epi’-memory? The role of epigenetics in nutritional programming, metabolic disease, aging and exercise. Aging Cell. 2016; 15(4): 603- 616. doi: 10.1111/acel.12486. Epub 2016 Apr 22. PMID: 27102569; PMCID: PMC4933662.
- McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol. 2009; 587(Pt 24): 5951-5958. doi: 10.1113/jphysiol.2009.181065. PMID:19884317; PMCID: PMC2808551.
- Li L. The Molecular Mechanism of Glucagon-Like Peptide-1 Therapy in Alzheimer’s Disease, Based on a Mechanistic Target of Rapamycin Pathway. CNS Drugs. 2017; 31(7): 535-549. doi: 10.1007/s40263-017-0431-2. PMID: 28540646.
- Zhang Z, Wang X, Zhang D, Liu Y, Li L. Geniposide-mediated protection against amyloid deposition and behavioral impairment correlates with downregulation of mTOR signaling and enhanced autophagy in a mouse model of Alzheimer’s disease. Aging (Albany NY). 2019; 11(2): 536-548. doi: 10.18632/aging.101759. PMID: 30684442; PMCID: PMC6366989.
- Kang EB, Cho JY. Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J Exerc Nutrition Biochem. 2015; 19(3): 199-209. doi: 10.5717/jenb.2015.15090806. Epub 2015 Sep 30. PMID: 26527331; PMCID: PMC4624121.
- Chen K, Zheng Y, Wei JA, Ouyang H, Huang X, Zhang F, et al. Exercise training improves motor skill learning via selective activation of mTOR. Sci Adv. 2019; 5(7): eaaw1888. doi: 10.1126/sciadv.aaw1888. PMID: 31281888; PMCID: PMC6609215.
- Brook MS, Wilkinson DJ, Mitchell WK, Lund JN, Phillips BE, Szewczyk NJ, et al. Synchronous deficits in cumulative muscle protein synthesis and ribosomal biogenesis underlie age-related anabolic resistance to exercise in humans. J Physiol. 2016; 594(24): 7399-7417. doi: 10.1113/JP272857. Epub 2016 Nov 7. PMID: 27654940; PMCID: PMC5157077.
- Wilkinson DJ, Piasecki M, Atherton PJ. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res Rev. 2018; 47: 123-132. doi: 10.1016/j.arr.2018.07.005. Epub 2018 Jul 23. PMID: 30048806; PMCID: PMC6202460.
- Fink J, Schoenfeld BJ, Nakazato K. The role of hormones in muscle hypertrophy. Phys Sportsmed. 2018; 46(1): 129-134. doi: 10.1080/00913847.2018.1406778. Epub 2017 Nov 25. PMID: 29172848.
- Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab. 2003; 285(1): E197-E205. doi: 10.1152/ajpendo.00370.2002. Epub 2003 Apr 1. PMID: 12670837.
- Liu XH, Wu Y, Yao S, Levine AC, Kirschenbaum A, Collier L, et al. Androgens up-regulate transcription of the Notch inhibitor Numb in C2C12 myoblasts via Wnt/β-catenin signaling to T cell factor elements in the Numb promoter. J Biol Chem. 2013; 288(25):17990- 17998. doi: 10.1074/jbc.M113.478487. Epub 2013 May 6. PMID: 23649620; PMCID: PMC3689944.
- Rahman F, Christian HC. Non-classical actions of testosterone: an update. Trends Endocrinol Metab. 2007; 18(10): 371-378. doi: 10.1016/j.tem.2007.09.004. Epub 2007 Nov 9. PMID: 17997105.
- Estrada M, Espinosa A, Müller M, Jaimovich E. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology. 2003; 144(8): 3586-3597. doi: 10.1210/ en.2002-0164. PMID: 12865341.
- McCall GE, Byrnes WC, Fleck SJ, Dickinson A, Kraemer WJ. Acute and chronic hormonal responses to resistance training designed to promote muscle hypertrophy. Can J Appl Physiol. 1999; 24(1): 96- 107. doi: 10.1139/h99-009. PMID: 9916184.
- Mangine GT, Hoffman JR, Gonzalez AM, Townsend JR, Wells AJ, Jajtner AR, et al. Exercise-Induced Hormone Elevations Are Related to Muscle Growth. J Strength Cond Res. 2017; 31(1): 45-53. doi: 10.1519/JSC.0000000000001491. PMID: 28005636.
- Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015; 14(1): 58-74. doi: 10.1038/nrd4467. PMID: 25549588.
- Tisdale MJ. The ubiquitin-proteasome pathway as a therapeutic target for muscle wasting. J Support Oncol. 2005; 3(3): 209-217. PMID: 15915823.
- Campos JC, Queliconi BB, Dourado PM, Cunha TF, Zambelli VO, Bechara LR, et al. Exercise training restores cardiac protein quality control in heart failure. PLoS One. 2012; 7(12): e52764. doi: 10.1371/ journal.pone.0052764. Epub 2012 Dec 27. PMID: 23300764; PMCID: PMC3531365.
- Bozi LH, Jannig PR, Rolim N, Voltarelli VA, Dourado PM, Wisløff U, et al. Aerobic exercise training rescues cardiac protein quality control and blunts endoplasmic reticulum stress in heart failure rats. J Cell Mol Med. 2016; 20(11): 2208-2212. doi: 10.1111/jcmm.12894. Epub 2016 Jun 16. PMID: 27305869; PMCID: PMC5082404.
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise- induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012; 481(7382): 511-515. doi: 10.1038/ nature10758. Erratum in: Nature. 2013 Nov 7:503(7474):146. PMID: 22258505; PMCID: PMC3518436.
- Witard OC, Jackman SR, Breen L, Smith K, Selby A, Tipton KD. Myofibrillar muscle protein synthesis rates subsequent to a meal in response to increasing doses of whey protein at rest and after resistance exercise. Am J Clin Nutr. 2014; 99(1): 86-95. doi: 10.3945/ ajcn.112.055517. Epub 2013 Nov 20. PMID: 24257722.
- Moore DR. Maximizing Post-exercise Anabolism: The Case for Relative Protein Intakes. Front Nutr. 2019; 6: 147. doi: 10.3389/ fnut.2019.00147. PMID: 31552263; PMCID: PMC6746967.
- Kraemer WJ, Ratamess NA, Hymer WC, Nindl BC, Fragala MS. Growth Hormone(s), Testosterone, Insulin-Like Growth Factors, and Cortisol: Roles and Integration for Cellular Development and Growth With Exercise. Front Endocrinol (Lausanne). 2020; 11: 33. doi: 10.3389/ fendo.2020.00033. PMID: 32158429; PMCID: PMC7052063.
- Zeng F, Zhao H, Liao J. Androgen interacts with exercise through the mTOR pathway to induce skeletal muscle hypertrophy. Biol Sport. 2017; 34(4): 313-321. doi: 10.5114/biolsport.2017.69818. Epub 2017 Sep 20. PMID: 29472733; PMCID: PMC5819476.
- Morton RW, Sato K, Gallaugher MPB, Oikawa SY, McNicholas PD, Fujita S, et al. Muscle Androgen Receptor Content but Not Systemic Hormones Is Associated With Resistance Training-Induced Skeletal Muscle Hypertrophy in Healthy, Young Men. Front Physiol. 2018; 9: 1373. doi: 10.3389/fphys.2018.01373. PMID: 30356739; PMCID: PMC6189473.
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016; 44(D1): D1251-D1257. doi: 10.1093/nar/gkv1003. Epub 2015 Oct 7. PMID: 26450961; PMCID: PMC4702768.
- Kudryavtseva AV, Krasnov GS, Dmitriev AA, Alekseev BY, Kardymon OL, Sadritdinova AF, et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016; 7(29): 44879- 44905. doi: 10.18632/oncotarget.9821. PMID: 27270647; PMCID: PMC5216692.
- Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016; 212(4): 379-387. doi: 10.1083/jcb.201511036. Epub 2016 Feb 8. PMID: 26858267; PMCID: PMC4754720.
- Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016; 61(5): 654-666. doi: 10.1016/j.molcel.2016.01.028. PMID: 26942670; PMCID: PMC4779179.
- Bornstein R, Gonzalez B, Johnson SC. Mitochondrial pathways in human health and aging. Mitochondrion. 2020; 54: 72-84. doi:10.1016/j.mito.2020.07.007. Epub 2020 Jul 30. PMID: 32738358;PMCID: PMC7508824.
- Memme JM, Erlich AT, Phukan G, Hood DA. Exercise and mitochondrial health. J Physiol. 2021; 599(3): 803-817. doi: 10.1113/JP278853.Epub 2019 Dec 9. PMID: 31674658.
- Robinson MM, Dasari S, Konopka AR, Johnson ML, Manjunatha S, Esponda RR, et al. Enhanced Protein Translation Underlies Improved Metabolic and Physical Adaptations to Different Exercise Training Modes in Young and Old Humans. Cell Metab. 2017; 25(3): 581-592. doi: 10.1016/j.cmet.2017.02.009. PMID: 28273480; PMCID: PMC5423095.
- CamposJC,Queliconi BB, BoziLHM, Bechara LRG, Dourado PMM, Andres AM, et al JCB. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy. 2017; 13(8): 1304-1317. doi: 10.1080/15548627.2017.1325062. Erratum in: Autophagy. 2024; 20(4): 979-981. doi: 10.1080/15548627.2024.2317238. PMID:28598232; PMCID: PMC5584854.
- Teng YC, Wang JY, Chi YH, Tsai TF. Exercise and the Cisd2 Prolongevity Gene: Two Promising Strategies to Delay the Aging of Skeletal Muscle. Int J Mol Sci. 2020; 21(23): 9059. doi: 10.3390/ijms21239059. PMID: 33260577; PMCID: PMC7731423.
- Yeh CH, Shen ZQ, Hsiung SY, Wu PC, Teng YC, Chou YJ, et al. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019; 17(10): e3000508. doi: 10.1371/journal. pbio.3000508. PMID: 31593566; PMCID: PMC6799937.
- Yokokawa T, Kido K, Suga T, Sase K, Isaka T, Hayashi T, et al. Exercise training increases CISD family protein expression in murine skeletal muscle and white adipose tissue. Biochem Biophys Res Commun. 2018; 506(3): 571-577. doi: 10.1016/j.bbrc.2018.10.101. Epub 2018Oct 23. PMID: 30366664.
- Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017; 313(5): H890-H895. doi: 10.1152/ajpheart.00416.2017. Epub 2017 Sep 29. PMID: 28971843; PMCID: PMC5792201.
- Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014; 15(7): 482-496. doi: 10.1038/ nrm3823. PMID: 24954210.
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positivecells shorten healthy lifespan. Nature. 2016; 530(7589): 184-189. doi: 10.1038/nature16932. Epub2016 Feb 3. PMID: 26840489; PMCID: PMC4845101.
- Schafer MJ, White TA, Evans G, Tonne JM, Verzosa GC, Stout MB, et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes. 2016; 65(6): 1606-1615. doi: 10.2337/db15-0291.Epub 2016 Mar 16. PMID: 26983960; PMCID: PMC4878429.
- Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017; 313(5): H890-H895. doi: 10.1152/ajpheart.00416.2017. Epub 2017 Sep 29. PMID: 28971843; PMCID: PMC5792201.
- Philippe M, Gatterer H, Burtscher M, Weinberger B, Keller M, Grubeck- Loebenstein B, et al. Concentric and Eccentric Endurance Exercise Reverse Hallmarks of T-Cell Senescence in Pre-diabetic Subjects. Front Physiol. 2019; 10: 684. doi: 10.3389/fphys.2019.00684. PMID:31214051; PMCID: PMC6558034.
- Jang Y, Kwon I, Cosio-Lima L, Wirth C, Vinci DM, Lee Y. Endurance Exercise Prevents Metabolic Distress-induced Senescence in the Hippocampus. Med Sci Sports Exerc. 2019; 51(10): 2012-2024. doi:10.1249/MSS.0000000000002011. PMID: 30998584.
- Cianflone E, Torella M, Biamonte F, De Angelis A, Urbanek K, Costanzo FS, et al. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells. 2020; 9(6): 1558. doi: 10.3390/ cells9061558. PMID: 32604861; PMCID: PMC7349658.
- Cable J, Fuchs E, Weissman I, Jasper H, Glass D, Rando TA, et al. Adult stem cells and regenerative medicine-a symposium report. Ann N Y Acad Sci. 2020; 1462(1): 27-36. doi: 10.1111/nyas.14243. Epub 2019Oct 26. PMID: 31655007; PMCID: PMC7135961.
- Tümpel S, Rudolph KL. Quiescence: Good and Bad of Stem CellAging. Trends Cell Biol. 2019; 29(8): 672-685. doi: 10.1016/j.tcb.2019.05.002. Epub 2019 Jun 24. PMID: 31248787.
- Vilas JM, Carneiro C, Da Silva-Álvarez S, Ferreirós A, González P, Gómez M, et al. Adult Sox2+ stem cell exhaustion in mice results in cellular senescence and premature aging. Aging Cell. 2018; 17(5): e12834. doi: 10.1111/acel.12834. Epub 2018 Aug 20. PMID: 30129215; PMCID: PMC6156495.
- Macaluso F, Myburgh KH. Current evidence that exercise can increase the number of adult stem cells. J Muscle Res Cell Motil. 2012; 33(3-4): 187-98. doi: 10.1007/s10974-012-9302-0. Epub 2012 Jun 7. PMID: 22673936.
- Bonsignore MR, Morici G, Santoro A, Pagano M, Cascio L, Bonanno A, et al. Circulating hematopoietic progenitor cells in runners. J Appl Physiol (1985). 2002; 93(5): 1691-1697. doi: 10.1152/japplphysiol. 00376.2002. PMID: 12381755.
- Brett JO, Arjona M, Ikeda M, Quarta M, de Morrée A, Egner IM, et al. Exercise rejuvenates quiescent skeletal muscle stem cells in old mice through restoration of Cyclin D1. Nat Metab. 2020; 2(4): 307- 317. doi: 10.1038/s42255-020-0190-0. Epub 2020 Apr 13. PMID: 32601609; PMCID: PMC7323974.
- Sen S. Adult Stem Cells: Beyond Regenerative Tool, More as a Bio-Marker in Obesity and Diabetes. Diabetes Metab J. 2019; 43(6): 744-751. doi: 10.4093/dmj.2019.0175. PMID: 31902144; PMCID: PMC6943270.
- Steiner S, Niessner A, Ziegler S, Richter B, Seidinger D, Pleiner J, et al. Endurance training increases the number of endothelial progenitor cells in patients with cardiovascular risk and coronary artery disease. Atherosclerosis. 2005; 181(2): 305-10. doi: 10.1016/j.atherosclerosis.2005.01.006. Epub 2005 Feb 12. PMID: 16039284.
- Jenkins NT, Witkowski S, Spangenburg EE, Hagberg JM. Effects of acute and chronic endurance exercise on intracellular nitric oxide in putative endothelial progenitor cells: role of NAPDH oxidase. Am J Physiol Heart Circ Physiol. 2009; 297(5): H1798-H1805. doi: 10.1152/ajpheart.00347.2009. Epub 2009 Aug 28. PMID: 19717732; PMCID: PMC2781362.
- Gensch C, Clever Y, Werner C, Hanhoun M, Böhm M, Laufs U. Regulation of endothelial progenitor cells by prostaglandin E1 via inhibition of apoptosis. J Mol Cell Cardiol. 2007; 42(3): 670-677. doi: 10.1016/j.yjmcc.2006.12.017. Epub 2007 Jan 9. PMID: 17291526.
- Schlager O, Giurgea A, Schuhfried O, Seidinger D, Hammer A, Gröger M, et al. Exercise training increases endothelial progenitor cells and decreases asymmetric dimethylarginine in peripheral arterial disease: a randomized controlled trial. Atherosclerosis. 2011; 217(1): 240-248. doi: 10.1016/j.atherosclerosis.2011.03.018. Epub 2011 Apr8. PMID: 21481871.
- Laufs U, Urhausen A, Werner N, Scharhag J, Heitz A, Kissner G, et al. Running exercise of different duration and intensity: effect on endothelial progenitor cells in healthy subjects. Eur J Cardiovasc Prev Rehabil. 2005; 12(4): 407-414. doi: 10.1097/01.hjr.00001748 23.87269.2e. PMID: 16079651.
- Hoetzer GL, Van Guilder GP, Irmiger HM, Keith RS, Stauffer BL, DeSouza CA. Aging, exercise, and endothelial progenitor cell clonogenic and migratory capacity in men. J Appl Physiol (1985). 2007; 102(3): 847-852. doi: 10.1152/japplphysiol.01183.2006. Epub2006 Dec 7. PMID: 17158243.
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006; 8(4): 315-317. doi: 10.1080/ 14653240600855905. PMID: 16923606.
- Schmidt A, Bierwirth S, Weber S, Platen P, Schinköthe T, Bloch W. Short intensive exercise increases the migratory activity of mesenchymal stem cells. Br J Sports Med. 2009; 43(3): 195-198. doi: 10.1136/bjsm.2007.043208. Epub 2007 Dec 10. PMID: 18070806.
- FrodermannV,RohdeD,CourtiesG,SevereN,SchlossMJ,AmatullahH,et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat Med. 2019; 25(11): 1761-1771. doi: 10.1038/s41591-019-0633-x. Epub 2019 Nov 7. PMID: 31700184; PMCID: PMC6858591.
- Shin MS, Park HK, Kim TW, Ji ES, Lee JM, Choi HS, et al. Neuroprotective Effects of Bone Marrow Stromal Cell Transplantation in Combination With Treadmill Exercise Following Traumatic Brain Injury. Int Neurourol J. 2016; 20(Suppl 1): S49-S56. doi: 10.5213/inj.1632616.308. Epub 2016 May 26. PMID: 27230460; PMCID: PMC4895912.
- Zhang YX, Yuan MZ, Cheng L, Lin LZ, Du HW, Chen RH, et al. Treadmill exercise enhances therapeutic potency of transplanted bone mesenchymal stem cells in cerebral ischemic rats via anti-apoptotic effects. BMC Neurosci. 2015; 16: 56. doi: 10.1186/s12868-015-0196- 9. PMID: 26342636; PMCID: PMC4560892.
- Li R, Liang L, Dou Y, Huang Z, Mo H, Wang Y, et al. Mechanical strain regulates osteogenic and adipogenic differentiation of bone marrow mesenchymal stem cells. Biomed Res Int. 2015; 2015: 873251. doi: 10.1155/2015/873251. Epub 2015 Apr 2. PMID: 25922842; PMCID: PMC4398939.
- Cianflone E, Torella M, Biamonte F, De Angelis A, Urbanek K, Costanzo FS, et al. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells. 2020; 9(6): 1558. doi: 10.3390/ cells9061558. PMID: 32604861; PMCID: PMC7349658.
- Wahl P, Brixius K, Bloch W. Exercise-induced stem cell activation and its implication for cardiovascular and skeletal muscle regeneration. Minim Invasive Ther Allied Technol. 2008; 17(2): 91-99. doi: 10.1080/13645700801969816. PMID: 18465444.
- Cameron-Smith D. Exercise and skeletal muscle gene expression. Clin Exp Pharmacol Physiol. 2002; 29(3): 209-213. doi: 10.1046/j.1440- 1681.2002.03621.x. PMID: 11906485.
- Macaluso F, Myburgh KH. Current evidence that exercise can increase the number of adult stem cells. J Muscle Res Cell Motil. 2012; 33(3-4): 187-198. doi: 10.1007/s10974-012-9302-0. Epub 2012 Jun 7. PMID: 22673936.
- O’Reilly C, McKay B, Phillips S, Tarnopolsky M, Parise G. Hepatocyte growth factor (HGF) and the satellite cell response following muscle lengthening contractions in humans. Muscle Nerve. 2008; 38(5): 1434-1442. doi: 10.1002/mus.21146. PMID: 18816607.
- Frodermann V, Rohde D, Courties G, Severe N, Schloss MJ, Amatullah H, et al M. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat Med. 2019; 25(11): 1761-1771. doi: 10.1038/ s41591-019-0633-x. Epub 2019 Nov 7. PMID: 31700184; PMCID: PMC6858591.
- Vonderwalde I, Kovacs-Litman A. Aerobic exercise promotes hippocampal neurogenesis through skeletal myofiber-derived vascular endothelial growth factor. J Physiol. 2018; 596(5): 761-763. doi: 10.1113/JP275582. Epub 2018 Jan 31. PMID: 29315566; PMCID: PMC5830424.
- Möbius-Winkler S, Hilberg T, Menzel K, Golla E, Burman A, Schuler G, et al. Time-dependent mobilization of circulating progenitor cells during strenuous exercise in healthy individuals. J Appl Physiol (1985). 2009; 107(6): 1943-1950. doi: 10.1152/ japplphysiol.00532.2009. Epub 2009 Oct 1. PMID: 19797690.
- Rich B, Scadeng M, Yamaguchi M, Wagner PD, Breen EC. Skeletal myofiber vascular endothelial growth factor is required for the exercise training-induced increase in dentate gyrus neuronal precursor cells. J Physiol. 2017; 595(17): 5931-5943. doi:10.1113/JP273994. Epub 2017 Jun 28. PMID: 28597506; PMCID:PMC5577548.
- Uysal N, Agilkaya S, Sisman AR, Camsari UM, Gencoglu C, Dayi A, et al. Exercise increases leptin levels correlated with IGF-1 in hippocampus and prefrontal cortex of adolescent male and female rats. J Chem Neuroanat. 2017; 81: 27-33. doi: 10.1016/j.jchemneu.2017.02.004. Epub 2017 Feb 4. PMID: 28179125.
- Quan H, Koltai E, Suzuki K, Aguiar AS Jr, Pinho R, Boldogh I, et al. Exercise, redox system and neurodegenerative diseases. Biochim Biophys Acta Mol Basis Dis. 2020; 1866(10): 165778. doi: 10.1016/j. bbadis.2020.165778. Epub 2020 Mar 25. PMID: 32222542.
- Broholm C, Pedersen BK. Leukaemia inhibitory factor--an exercise- induced myokine. Exerc Immunol Rev. 2010; 16: 77-85.
- Farup J, De Lisio M, Rahbek SK, Bjerre J, Vendelbo MH, Boppart MD, et al. Pericyte response to contraction mode-specific resistance exercise training in human skeletal muscle. J Appl Physiol (1985). 2015; 119(10): 1053-1063. doi: 10.1152/japplphysiol.01108.2014. Epub 2015 Sep 24. PMID: 26404620.
- Fafián-Labora JA, O’Loghlen A. Classical and Nonclassical Intercellular Communication in Senescence and Ageing. Trends Cell Biol. 2020; 30(8): 628-639. doi: 10.1016/j.tcb.2020.05.003. Epub 2020 Jun 3. PMID: 32505550.
- Boucher MJ, Jean D, Vézina A, Rivard N. Dual role of MEK/ERK signaling in senescence and transformation of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004; 286(5): G736-G746. doi: 10.1152/ajpgi.00453.2003. Epub 2003 Dec 30. PMID: 14701721.
- Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: Insights into the pathophysiology of lung diseases. Mol Aspects Med. 2018; 60: 92-103. doi: 10.1016/j.mam.2017.11.005. Epub 2017 Nov 20. PMID: 29146100.
- Schwab N, Grenier K, Hazrati LN. DNA repair deficiency and senescence in concussed professional athletes involved in contact sports. Acta Neuropathol Commun. 2019; 7(1): 182. doi: 10.1186/ s40478-019-0822-3. PMID: 31727161; PMCID: PMC6857343.
- Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence- associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5: 99-118. doi: 10.1146/annurev- pathol-121808-102144. PMID: 20078217; PMCID: PMC4166495.
- Ha DH, Kim HK, Lee J, Kwon HH, Park GH, Yang SH, et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells. 2020; 9(5): 1157. doi: 10.3390/cells9051157. PMID: 32392899; PMCID: PMC7290908.
- Borghesan M, Fafián-Labora J, Eleftheriadou O, Carpintero- Fernández P, Paez-Ribes M, Vizcay-Barrena G, et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019; 27(13): 3956-3971.e6. doi: 10.1016/j. celrep.2019.05.095. PMID: 31242426; PMCID: PMC6613042.
- Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun. 2017; 8: 15729. doi: 10.1038/ncomms15728. PMID: 28585531; PMCID: PMC5467215.
- Alibhai FJ, Lim F, Yeganeh A, DiStefano PV, Binesh-Marvasti T, Belfiore A, et al. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell. 2020; 19(3): e13103. doi: 10.1111/acel.13103. Epub 2020 Jan 21. PMID: 31960578; PMCID: PMC7059145.
- Mobarak H, Heidarpour M, Lolicato F, Nouri M, Rahbarghazi R, Mahdipour M. Physiological impact of extracellular vesicles on female reproductive system; highlights to possible restorative effects on female age-related fertility. Biofactors. 2019; 45(3): 293-303. doi: 10.1002/biof.1497. Epub 2019 Feb 20. PMID: 30788863.
- Willis GR, Fernandez-Gonzalez A, Reis M, Yeung V, Liu X, Ericsson M, et al. Mesenchymal stromal cell-derived small extracellular vesicles restore lung architecture and improve exercise capacity in a model of neonatal hyperoxia-induced lung injury. J Extracell Vesicles. 2020; 9(1): 1790874. doi: 10.1080/20013078.2020.1790874. PMID: 32939235; PMCID: PMC7480622.
- Estébanez B, Jiménez-Pavón D, Huang CJ, Cuevas MJ, González- Gallego J. Effects of exercise on exosome release and cargo in in vivo and ex vivo models: A systematic review. J Cell Physiol. 2021; 236(5): 3336-3353. doi: 10.1002/jcp.30094. Epub 2020 Oct 9. PMID: 33037627.









































































































































{kind=link}