Metabolic Pathways of Endogenous Formaldehyde
- 1. National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Academician V.I.Kulakov, Russia
- 2. Limited Liability Company “SARBIOTEKH”, Russia
- 3. Russian research institute of health (RIH), Russia
- 4. World Wide Medical Assistance 6317 Switzerland/ Obervill, Zug, Fushtok
Abstract
It is generally accepted that formaldehyde (FA) is a toxic substance. It is formed in various demethylation and transmethylation reactions. And there is an intracellular system for its detoxification. Meanwhile, its effect on the cell causes a different response: apoptosis, proliferation, differentiation and depends on the concentration of FA. Evolutionarily, FA is a component of the one-carbon transport system. Oxidation of the methyl group in the folate cycle is a proton donor for the reduction of NADP+. In the cell, FA spontaneously non-enzymatically binds to pterin, arginine, glutathione, which determines its participation in the metabolism of purines, nitric oxide and polyamines, and in the redox system. This review describes a number of metabolic pathways that are linked into a single system thanks to FA. FA is the main source of formate in the cell and closely interacts with arginine metabolic pathways, influencing the synthesis of nitric oxide and polyamines. The possible role of formaldehyde dehydrogenase (ADH5) in the regulation of intracellular pH is also considered.
Citation
Tereshina EV, Laskavy VN, Alekseev AA, Polyanina TI, Sukhikh GT (2025) Metabolic Pathways of Endogenous Formaldehyde. J Pharmacol Clin Toxicol 13(1):1189.
Keywords
• Formaldehyde; Metabolism; Nitric oxide; Polyamines
INTRODUCTION
The biogenic carbon redox cycle is carried out by two types of biosystems – methanogens and methanotrophs. Methanogenic archaea are strictly anaerobic Euryarchaeota that reduce carbon dioxide to methane. They use molecular hydrogen, formate, methanol, methylamines and acetate as proton donors [1]. The reduction of CO2 occurs sequentially along the line “carbon dioxide-formate-formaldehyde-methanol-methane”. The formyl group is transferred from methanofuran to a pterin-containing compound, tetrahydrosarcinapterin (H4MPT), an analogue of pterin-containing tetrahydrofolic acid (THF). One-carbon compounds (C1) bind to pterin. The second half of the carbon redox cycle is the oxidation of methane by methylotrophs. Methylotrophs are a heterogeneous group of microorganisms, represented by obligate and facultative bacteria. In these bacteria, methane is oxidized enzymatically to formaldehyde (FA), which combines with two C1-compound carriers: H4MPT to form methylene-H4MPT and THF to form methyleneTHF. In these microorganisms, FA also enters metabolic pathways through the ribulose monophosphate pathway, in which glyceraldehyde-3-phosphate is formed, and through the serine pathway, in which methylene-THF interacts with glycine to form serine [2]. In mammals, the pathway for transporting C1 bound to THF is preserved, and methylene-THF is possibly involved in the synthesis of serine from glycine. Spontaneous binding of FA to THF has been shown in the pH range of 5.4-8.5. In pig liver extracts, FA was incorporated into serine in the pH range of 7.7-8.0 [3]. Further evolution followed the line of methylotrophs, where FA is oxidized to formate.
SOURCES OF ENDOGENOUS FORMALDEHYDE
FA is a by-product of various transmethylation [4] and demethylation reactions, including histones that contain methyl lysines [5]. Histone demethylation is irreversible. As a result of the action of the enzyme LSD 1 (lysinespecific demethylase 1), one molecule of FA is released from monomethyl lysine, two from dimethyl lysine, and three from trimethyl lysine [6].
FA is released during the demethylation of dimethylglycine (sarcosine is formed) and during the synthesis of glycine from sarcosine. Serine and glycine are the main suppliers of methyl groups for the folate cycle [7]. As a result of the demethylation of serine with the participation of THF, 5,10-methylene-THF (CH2-THF) and glycine are formed, which breaks down in the glycinecleavage system (GCS) also with the formation of CH2-THF [8].
Kikuchi et al.[9] found that FA can be released into GCS in the absence of THF. But then spontaneously, nonenzymatically, reacts with THF to form CH2-THF [10]. However, CH2-THF is highly unstable at pH 7.5 and also spontaneously decomposes into FA and THF, while THF returns to GCS and the glycine degradation cycle does not stop [11]. Jägerstad M. et al. [12], reported that CH2-THF can spontaneously dissociate into THF and FA at both physiological and acidic pH values, and is stabilized only at pH > 8.0 or in the presence of a very large excess of FA.
In an acidic environment, THF spontaneously undergoes oxidative degradation. THF degrades to form N-(4-aminobenzoyl)-L-glutamic acid, FA, and pterin, and as the environment becomes more acidic, FA becomes more abundant [13]. When THF interacts with FA to form CH2-THF, its degradation is slowed down. It is assumed that under physiological conditions, CH2-THF still remains stable and does not degrade, and it is more stable than THF [10]. When the intracellular FA level is high, THF is stabilized and forms CH2-THF, which simultaneously reduces FA production and its content in the cell. CH2-THF is consumed in the synthesis of thymidine and methionine. When the FA level is low, some unreacted THF is degraded and FA is produced. In this case, THF itself is a source of methyl group for the folate cycle. This means that the folate cycle can exist as a self-sustaining system, independent of the associated metabolic pathways and enzyme action, which is regulated by the concentration of PA and the pH of the environment.
CONNECTION OF METABOLIC PATHWAYS
Dihydrofolate reductase reduces dietary folate to dihydrofolic acid, which is then reduced by the same enzyme to THF. The enzyme cofactor is NADP+. Methyl groups are added to THF at N5 and/or N10 positions and have different oxidative status, which corresponds to methanol (5-methyl THF, CH3-THF), formaldehyde (5,10-methylene THF, CH2-THF) and formate (5,10-methenyl THF, CH-THF; 10-formyl THF, 10-CHO-THF and 5-formyl THF, 5-CHOTHF). n fact, the sequence of oxidation of the methyl group is built in accordance with the stages of methane oxidation: “methane-methanol-formaldehyde-formic acid-carbon dioxide”. Oxidation of the methyl group is the main proton donor (source of NADPH) in the conjugated redox system [14]. Fan et al. [15], performed a quantitative flux analysis, which revealed that NADPH is produced in the cell mainly by oxidation of cytoplasmic CH2-THF to CHO-THF.
In the cytoplasm, the enzyme serine hydroxymethyltransferase 1 (SHMT1) is active, which transfers the methyl group to glycine and, in fact, cannot participate in the formation of CH2-THF. In this case, FA can be a donor of the methyl group for binding to THF in the cytoplasm. FA may thus link the folate cycle with the cycle of glycine formation from serine via the serine-ethanolamine-choline-betaine aldehyde-betainedimethylglycine-sarcosine-glycine pathway. It is possible that FA released during demethylation of dimethylglycine and sarcosine is also involved in serine formation. Ethanolamine is a component of phosphatidylcholine, and choline is a component of phosphatidylcholine.
THF (Figure 1), is used as a carrier of intermediate products of the methyl group oxidation chain, thereby providing 1C-compounds for various metabolic pathways, including the biosynthesis of polyamines and phospholipids, as well as in genome methylation reactions. The metabolism of 1C-compounds is key for proliferating cells, as it provides components necessary for the synthesis of nucleic acids [16].
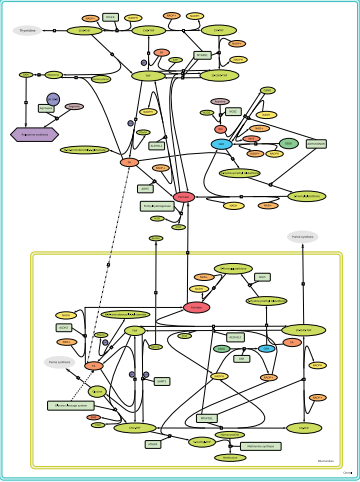
Figure 1: Coupling of metabolic pathways of formate formation in the cytoplasm and mitochondria
The folate cycle is coupled with the methionine cycle and is localized in the cytoplasm, mitochondria and nucleus [16]. CH2-THF is a substrate for methylenetetrahydrofolate reductase (MTHFR), which irreversibly converts CH2-THF to 5-methyltetrahydrofolate (CH3-THF), which plays a key role in methionine metabolism.
Thymidylate synthase (TYMS) uses CH2-THP as a methyl donor and methylates deoxyuridine monophosphate to form thymidine monophosphate [17]. Glycine, formed in the mitochondria in the reaction with SHMT2, is needed for the synthesis of purines, as well as glutathione (together with cysteine and glutamate). In addition to glycine, 10-CHO-THF is also needed for the synthesis of purines. The formyl group of 10-CHO-THF is used by phosphoribosylaminoimidazolecarboxamide formyltransferase and phosphoribosylglycinamide formyltransferase, which insert it into positions 2 and 8 of the purine ring carbons. With the participation of ATP, FA can non-enzymatically interact with THF to form 10-CHOTHF.
5-CHO-THF is not used as a cofactor in folatedependent synthesis reactions and is considered a storage form of THF in dormant cells [18]. The enzyme methenyltetrahydrofolate synthetase (MTHFS) irreversibly converts 5-CHO-THF to CH-THF using ATP. CH-THF can non-enzymatically produce two forms of formyl THF: 5-CHO-THF and 10-CHO-THF [19].
1C-METABOLISM AND FOLATE CYCLE
In the mitochondria, CH2-THF is formed by demethylation of serine, as well as in GSC. In the cytoplasm,the source of CH2-THF may be the non-enzymatic interaction of THF and FA. CH2-THF is enzymatically and sequentially converted into CH-THF and 10-CHOTHF. 10-CHO-THF is used in the synthesis of purines or is a source of formate and THF, which is returned to the folate cycle. Formate is oxidized with the release of carbon dioxide. The reversible oxidation of CH2-THF to CHO-THF is catalyzed by the enzyme methylenetetrahydrofolate dehydrogenase (MTHFD). There are two isoforms of this enzyme: cytoplasmic MTHFD1 and mitochondrial MTHFD2. MTHFD1 is a trifunctional enzyme, it exhibits in addition to CH2-THF dehydrogenase activity also CH2- THF cyclohydrolase and 10-CHO-THF synthetase activity, which uses NADP+ as a cofactor, i.e. NADPH is formed in this reaction. MTHFD1 catalyzes not only the reversible two-step conversion of CH2-THF to CHO-THF [20], but also the ATP-dependent synthesis of CHO-THF from formate and THF.
MTHFD2 is a bifunctional enzyme, it exhibits only dehydrogenase and cyclohydrolase activities and uses NAD+ as a cofactor. However, knockdown of MTHFD2 showed that this enzyme is also involved in the production of NADPH, although it preferentially uses NAD+ [21]. A mitochondrial analogue of MTHFD2, MTHFD2L, has also been identified, which uses NADP+ as a cofactor. It is expressed at a low level in the early stages of mouse embryo development, and its expression increases by day 10. It is then found in all organs of the adult animal, most prominently in the lungs and brain [22,23]. Mammalian fibroblasts deficient in MTHFD2 were glycine auxotrophs due to accumulation of CH2-THF, a substrate of MTHFD2, which inhibits SHMT2 and blocks glycine formation [24]. Expression of GCS genes is accompanied by increased levels of folate pathway enzymes, including MTHFD2.
Mitochondrial folate enzymes produce formate to a greater extent than cytoplasmic ones. This is due to the mitochondrial enzyme MTHFD1L, which exhibits only 10-CHO-THF synthetase activity, supplying the substrate for nucleotide synthesis [25]. However, it catalyzes the reverse reaction with greater efficiency, releasing formate from 10-CHO-THF [26].
The formyl group in 10-CHO-THF can be oxidized with the release of CO2 and the simultaneous reduction of NADP+ to NADPH in both the cytoplasm and mitochondria [27]. The oxidative degradation of 10-CHO-THF to THF and CO2 is catalyzed by the enzyme 10-formyltetrahydrofolate dehydrogenase (10-FDH). Since 10-CHO-THF is a substrate for purine biosynthesis reactions, it was suggested that by reducing the intracellular purine content, 10-FDH inhibits cell proliferation [28,29]. At the same time, it appears to regulate the 1C content of THF metabolites, as this is the last reaction of the folate pathway. The reaction product, CO2, leaves the cell. This enzyme could be considered in terms of regulating formate concentration in the cell [30], if there were no pathways for direct oxidation of FA to formate.
10-FDH contains pentaglutamyl-THF as a tightly bound non-catalytic cofactor. The 10-FDH gene is very similar in nucleotide sequence to the genes of enzymes of the aldehyde dehydrogenase family, so its other name is aldehyde dehydrogenase (ALDH).
There are two isoforms of the enzyme: cytosolic ALDH1L1 and mitochondrial ALDH1L2 [27,30]. ALDH1L1 is the most abundant cytosolic enzyme of 1C metabolism, its amount reaches 1.2% of the total protein in rat liver [27,30,31]. ALDH1L1 is expressed in many tissues at levels several times higher than ALDH1L2 expression [30]. The contribution of this enzyme to the cytosolic pool of intracellular NADPH has not yet been established. In mitochondria, NADPH production in 1C metabolism is coupled with the enzymatic activity of ALDH1L2 [15,32].
It has been suggested that this reaction is the main supplier of NADPH to mitochondria [33]. ALDH1L2 is the product of the ALDH1L2 gene. It has approximately 74% similarity with the cytosolic 10-FDH gene, and the identity of the dehydrogenase domain reaches 79% [34]. Unlike its homologue, ALDH1L2 is expressed in malignant tumors and is involved in the regulation of metastasis [35].
Since the sequential oxidation reactions of THF metabolites, including 10-CHO-THF, are reversible, it is believed that 10-FDH may be a major regulator of the folate cycle and related metabolic pathways.
FA is one of the sources of formate in the cell [36]. When mammalian cells were incubated in a medium containing physiological concentrations of FA (20-40 mM), it was the source of formation of 10% to 50% of all cellular formate [37]. In the cytosol, the enzyme formate-THF ligase, the activity of the trifunctional enzyme MTHFD1, binds it to THF in an ATP-dependent reaction to form CHO-THF [38]. It is unknown under what conditions the enzymatic and non-enzymatic binding of FA to THF using ATP occurs.
FORMALDEHYDE DETOXIFICATION
FA spontaneously reacts with sulfhydryl groups of cysteine, particularly in reduced glutathione (GSH). The cytosolic zinc-containing enzyme alcohol dehydrogenase class 5 (ADH5), also known as alcohol dehydrogenase class III (ADH3) and formaldehyde dehydrogenase (FALDH), oxidizes the resulting hydroxymethyl-GSH (HMGSH) using NAD+ to formyl-GSH [39]. Formyl-GSH hydrolase (esterase D hydrolase activity) then hydrolyzes formyl-GSH to formate and GSH [40]. Formate binds to THF (CH-THF) or is oxidized to CO2. FA, which in an acidic environment (pH < 7.4) is formed as a result of the breakdown of THF and CH2-THF, at pH> 7.5 spontaneously interacts with both THF and GSH. GSH and THF compete for binding to FA [41,42]. However, THF has been shown to be significantly more efficient at capturing FA than GSH, indicating that CH2-THF can persist in a glutathione-rich environment [43]. The folate cycle and the glutathione system are coupled not only at the level of NADPH formation, which is used to reduce oxidized GSSG to GSH, but also at the level of maintaining certain FA concentrations in the cell, including through the formation of HMGSH. The interaction of FA with GSH, in this case, should not be considered as its detoxification.
Metabolomic analysis shows that formate, as a product of FA oxidation associated with glutathione, is actively involved in DNA and ATP synthesis, since the radioactive label in nucleotides did not appear in ADH5-deficient cells [44] (Km = 0.12–6.5 μM). However, the culture of chicken DT40 cells lacking the ADH5 gene can grow normally, and their sensitivity to exogenous FA does not differ from that of wild-type cells [44]. The lifespan (LS) of Adh5−/− mice did not differ from that of wild-type animals [45]. Moreover, Adh5−/− mice of both sexes were born without defects and developed normally [46]. It was suggested that there is another metabolic pathway of FA not associated with ADH5. FA can be oxidized to formate by mitochondrial aldehyde dehydrogenase 2 (ALDH2, Km = 170–400 μM) [47]. ALDH2 with a relatively low Km can be considered a compensatory enzyme in relation to ADH5.
The main substrate for ALDH2 is acetaldehyde, the cofactor is NAD+. ALDH2 oxidizes acetaldehyde to acetate and is important in ethanol metabolism [48]. Mice knocked out in two genes, mitochondrial ALDH2 and cytoplasmic ADH5, had a very short lifespan, developed leukemia, had profoundly impaired hematopoiesis, and had decreased levels of hematopoietic stem cells and lymphoid progenitors, which led to the loss of acquired immunity [49]. These mice had an 11-fold increase in blood FA levels, which correlated with both FA-induced DNA damage and mutagenesis similar to that observed in human cancer cells [50]. Decreased ALDH2 activity is associated with decreased oxygen consumption rate. ALDH2-deficient mice had decreased mitochondrial respiration, despite unchanged expression of electron transport chain (ETC) proteins [51]. This may be due to inhibition of ETC complex 4 by nitric oxide.
The effect of FA on the cell is dose-dependent. At a concentration of 1.0–10 μg/ml of analytically pure FA, apoptosis was observed in the culture of cancer and endothelial cells, and at a concentration of 0.1–0.01 μg/ ml, cell proliferation was observed [52,53]. When rat hepatocytes were incubated in a medium with a low concentration of FA, a dose-dependent decrease in the membrane potential of mitochondria was observed, accompanied by the formation of ROS. The content of GSH also decreased dose-dependently [54]
Thus, manipulations that change the intracellular FA content can affect the functioning of the folate cycle and cell proliferation. In normal cells, the intracellular pH is <7.4, and then the folate cycle does not function. It is only triggered in proliferating cells at alkaline intracellular pH. How can formate formation during a functioning folate cycle affect intracellular pH?
METHIONINE CYCLE AND POLYAMINES
Ectopic expression of 10-FDH results in decreased levels of CH3-THF and S-adenosyl methionine (SAM) [55]. MTHFR, with the participation of NADPH, irreversibly reduces CH2-THF to CH3-THF, which couples the folate cycle with the methionine cycle. CH3-THF is a methyl group donor for methionine synthase, which catalyzes the synthesis of methionine from homocysteine. Of all the metabolites of the folate cycle, CH3-THF is the one that is formed in the greatest quantity [56]. In mammals, methionine synthesis is the only reaction involving CH3- THF [57]. MTHFR is allosterically inhibited by SAM [58,59]. The methionine cycle supplies methyl groups to methylases that methylate DNA. CH3-THF allosterically inhibits glycine N-methyltransferase (GNMT) [60]. GNMT catalyzes the conversion of SAM to S-adenosylhomocysteine (SAH) and plays an important role in maintaining the SAM/SAH ratio [61]. GNMT activity is regulated by the concentrations of both SAM, which inhibits MTHFR and the formation of CH3-THF, and CH3-THF itself [62]. Deletion of the GNMT gene results in a significant increase in SAM levels and changes in methylation status in vivo. GNMT is one of the factors regulating proliferation and carcinogenesis [63,64]. Demethylation of SAM in the methionine cycle releases FA [65]. At the same time, FA methylates homocysteine to form methionine. SAM is involved in the synthesis of polyamines. Arginase is active at alkaline pH (7.5-8.0) and functions in proliferating cells.
NITRIC OXIDE AS A COMPONENT OF THE CONJUGATED REDOX SYSTEM
Nitric oxide (NO) exhibits two opposing effects: on the one hand, it inhibits proliferation, on the other, it activates it. This paradox is explained by its concentrations that were used in different experiments. High concentrations of NO suppress, while low concentrations activate the proliferation of both normal and tumor cells [66]. In the microenvironment of tumor cells, the content of NO is recorded in nanomolar (nmol/L) quantities. It is these concentrations that support carcinogenesis.
In the search for intracellular targets of nitric oxide that induce proliferation in human breast cancer cell lines MDAMB-231 and MCF-7, the NO donor DETA/NO, characterized by a slow release of NO and a half-life of about 20 hours, was used. NO in nanomolar concentrations significantly increased protein synthesis, primarily the translation of cyclin D1, without affecting the level of their mRNA [67]. When using DETA/NO in a culture of hematopoietic stem and progenitor cells, its biphasic effect was revealed. At low concentrations DETA/NO activated classical NO signaling and proliferation, at high concentrations DETA/ NO slowed cell division and initiated their differentiation. Exposure of embryonic stem cells to low concentrations of NO (from 2 to 20 μmol/l) can slow differentiation [68]. In another range of low concentrations (10 - 50 μM/l), the NO donor protected mouse bone marrow stromal cells from spontaneous apoptosis [69]. The arrest of keratinocyte proliferation under the influence of NO occurs simultaneously with an increase in the activity of cytosolic superoxide dismutase 1 (SOD1) [70]. In mitochondria, SOD2 neutralizes superoxide anion, increasing NO production, which is observed in fibrosarcoma cells [71]. NO is involved in proliferation, including through pathways associated with polyamine metabolism [72].
ETC components have different sensitivities to NO inhibition. At concentrations below 0.2 μM, NO reversibly inhibits COX, controlling mitochondrial respiration; in the range of 0.3–0.5 μM, it inhibits electron transfer between cytochromes b and c1 [73]. With relatively long-term exposure at a concentration of 0.5–1 μM NO, the activity of NADH dehydrogenase complex I is selectively inhibited in intact cells [74].
Extracellular pH affects the production of NO and hydroxyl radical in hepatocytes. As a result of acidification of the medium (pH 7.0), there is a significant increase in OH radicals, cell damage and a sharp decrease in glutathione content. Incubation at physiological pH 7.4 led to an increase in NO synthesis, and at pH 7.8 - to a significant increase [75]. Meanwhile, inducible NO synthase (iNOS) is active at acidic pH (7.0-7.4) and utilizes NADPH, the oxidation product of the methyl group of CH2-THF and formate in the folate cycle. At physiological (pH 7.4) and alkaline (pH 7.8) pH values, a decrease in ONOO- formation correlated with a decrease in superoxide anion formation and an increase in GSH levels [76]. Thus, at acidic pH values, NO interacts with superoxide anion to form ONOO-, and at alkaline pH values, with GSH. In many normal and cancer cells, increased GSH levels are associated with a proliferative response [77]. Elevated GSH levels have been observed in a variety of cancers including breast, ovarian, lung, head and neck cancers [78]. There is a direct correlation between GSH levels, proliferation and metastasis [79].
NO competes with FA for binding to GSH. ADH5 (FALDH) uses not only HMGSH but also nitrosoglutathione (GSNO) as a substrate, reducing it to glutathione and ammonia. GSNO is formed non-enzymatically by the interaction of ONOO- with cysteine in glutathione. This catalytic activity has given FALDH another name – nitrosoglutathione reductase (GSNOR). It can restore nitrosylated cysteine residues not only in glutathione but also in proteins [80]. Attention has been drawn to this enzyme since it became clear that NO is a signaling molecule involved in a wide variety of physiological processes [81].
GSNOR is a cytosolic zinc-containing enzyme that functions as a dimer. Loss of GSNOR activity leads to the development of nitrosative stress [82]. The rate of GSNO catalysis is 20 times higher than the rate of HMGSH catalysis [83]. The products of reactions catalyzed by GSNOR are ammonia (in the case of GSNO) and formate (in the case of HMGSH), which can affect intracellular pH depending on the concentrations of NO and FA [84]. It can be speculated that ADH5 in the coupled redox system can serve as a basal regulator of intracellular pH by releasing formate and ammonia.
NOS AND FORMALDEHYDE
Both excess and deficiency of arginine in the medium affect cell proliferation. Deficiency of arginine causes apoptosis [85]. In cells of some types of cancer, a combined activation of iNOS and arginine synthase is observed [86,87]. Arginine is a substrate for the synthesis of NO and polyamines. It is methylated at the guanidine group, yielding hydroxymethyl and methyl derivatives. Methylarginines inhibit NOS [88]. Arginine is methylated by SAM and FA [89].
The methyl group of SAM is transferred to the guanidine nitrogen of arginine residues of proteins. This transfer is catalyzed by SAM- or AdoMet-dependent methyltransferases PRMT (protein N-arginine methyltransferases) [90,91]. In mammalian cells, the main methyl group carriers are PRMT1 and PRMT5. They methylate almost all arginines in proteins. However, in human cells and yeast, about 80% of all methyltransferase activity is accounted for by PRMT1 [92]. PRMTs are regulated by kinases. Thus, phosphorylation of CARM1 (PRMT4) leads to a decrease in its activity [93]. In proteins, arginine can be methylated in three different ways. During proteolysis in proteasomes or lysosomes, small molecules of NG-monomethyl-l-arginine (l-NMMA, MMA), NG, NG-dimethylarginine (asymmetric dimethylarginine, ADMA), and NG, NG’-dimethylarginine (symmetric dimethylarginine, SDMA) are released from proteins. ADMA is obtained by methylation of arginine by methylase PRMT1, SDMA – by methylation by methylase PRMT2.
ADMA and l-NMMA are the most potent NOS inhibitors. They inhibit all three isoforms almost equally: iNOS, eNOS (endothelial), nNOS (neuronal). Due to its high concentration, ADMA inhibits NOS more actively than l-NMMA. ADMA competes with arginine not only for binding to NOS, but also for transport into the cell [94]. The Na+ transport system Y+ CAT (cation arginine transport), common to all N-methylarginines and arginine, transports ADMA and SDMA with varying efficiency [95]. The concentration of arginine in blood plasma is ~ 100 to 200 µM, the concentration of ADMA is significantly lower: 0.3– 1.0 µM [96]. However, the intracellular content of ADMA is much higher than the extracellular content: 5-20 times [97]. CAT probably does not ensure equilibrium between intracellular and extracellular ADMA levels. Intracellular ADMA can inhibit CAT by limiting both ADMA efflux and arginine influx [96]. Access of extracellular arginine to NOS is facilitated by the enzyme’s close association with the arginine Y+ transporter. Activation or inhibition of NOS thus depends on the L-arginine/ADMA concentration ratio [98,99]. SDMA has only one methyl group on each nitrogen of the guanidine group. This methylarginine is a structural isomer of ADMA. However, SDMA does not inhibit NO, but deactivates SAT and limits the bioavailability of arginine, thereby indirectly affecting NOS activity [100].
SDMA is excreted from the bloodstream in the urine [101], and ADMA and L-NMMA are catabolized by DDAH 1 (Dimethylarginine Dimethylaminohydrolase, two isoforms DDAH 1 and DDAH 2) to form dimethylamine and citrulline [96]. Part of the dimethylamine is demethylated to form methylamine and FA. Oxidative deamination of methylamine produces three products: ammonia, FA, and hydrogen peroxide [102]. Both isoenzymes have a higher affinity for ADMA than for L-NMMA [102,103].
DDAH is a semicarbazide-sensitive amine oxidase. It belongs to a group of copper-containing amine oxidases that are inhibited by semicarbazide. DDAH is a homodimeric transmembrane glycoprotein that has a large extracellular domain with a catalytic center containing Cu2+. The enzyme cofactor is 6-hydroxydopaquinone. The presence of Cu2+ in the active site indicates the possibility of direct inhibition of this enzyme by NO. S-nitrosylation of cysteines in the active site of DDAH (Cys-249) by nitric oxide also inhibits the enzyme. When ADMA accumulates, it in turn blocks NO synthesis. At the same time, accumulating methylamine and dimethylamine can enhance NO synthesis. Methylarginines can also regulate the formation of superoxide anion by NO synthases [103,104]. When BH4 is oxidized to BH2, ADMA stimulates the production of ROS, which in turn increases the concentration of intracellular ADMA. At the same time, ONOO- increases the synthesis of free FA through the modification of proteins by oxidizing their methionine residues into methionine sulfoxide. After release from proteins as a result of proteolysis, methionine sulfoxide, under conditions of hyperproduction of NO and ROS, undergoes further oxidation to form FA [105].
The guanidine group of arginine can bind one, two, or three FA molecules, resulting in the formation of mono-, di-, and tri-hydroxymethylated derivatives of arginine [106]. The hydroxymethyl groups are bound to the guanidine group reversibly, so that arginine can either accept or donate an FA molecule. Hullán et al. [107], noted that NG-hydroxymethylated arginine regulates the level of endogenous FA. Arginine interacts with FA both in free form and as part of proteins. And transfers it to the folate cycle and to the synthesis of thymidine. Hydroxymethylated derivatives of arginine are relatively stable compounds. They can serve as a reservoir and carriers of FA [4].
NG-hydroxymethylarginine dose-dependently and significantly inhibited the proliferation of HT-29 colon carcinoma cells, P-388 mouse lymphoma cells, PC-3 prostate carcinoma cells, and K-562 human erythroleukemia cells in culture [108]. ?In vivo experiments were performed by intraperitoneally administering 400 mg/kg hydroxymethylated arginine to mice with transplantable Ehrlich ascites carcinoma daily. Complete cessation of tumor growth was achieved after 10 days [109].
ARGINASE AND FORMALDEHYDE
Arginine is a substrate for both NOS and arginase. Increased expression of arginase has been reported in many types of human cancer [110,111]. Polyamines are small cationic molecules that are involved in cell proliferation and differentiation at millimolar concentrations [112,113], regulation of signal transduction, expression of cell cycle genes [114,115]. Increased intracellular polyamine content is associated with tumor development [113]. At the same time, intracellular accumulation of polyamines induces apoptosis [116], through activation of caspases [117].
The optimal pH for arginase is 9.0–9.5 [118]. An intermediate in nitric oxide synthesis, N-hydroxy-L-arginine (NOHA), is an arginase inhibitor [119-121]. S-nitrosylation of arginase cysteines is a post-translational modification of the enzyme, which does not reduce, but increases its activity [122]. Inhibition of arginase 1 stimulates NO production, which reduces polyamine synthesis by shutting down ornithine decarboxylase (ODC), the first enzyme in the metabolic pathway for polyamine synthesis from ornithine [123]. NO inhibits ODC by S-nitrosylation of cysteine residues in its catalytic center, thereby blocking polyamine production and cell proliferation [124]. At the same time, in cancer cells, ODC activity is constantly elevated [125,126]. In colorectal cancer cells, the content of polyamines, the activity of ODC and another enzyme of their synthesis, S-adenosylmethionine decarboxylase, is increased by 3 times compared to intact cells [127,128]. In the synthesis of methionine from homocysteine, CH3- THF and FA directly are involved. Thus, the synthesis of polyamines is associated with the synthesis of nucleotides, and with FA both through CH2-THF and directly.
Polyamines regulate MYC expression, whereas c-Myc regulates ODC expression at the transcriptional level [129,130]. ODC translation is affected by ammonia accumulation, significantly reducing it. Ammonia inhibits polyamine biosynthesis and cell proliferation.
In normal cells, the content of reduced glutathione is 98% of its total amount [131]. With an increase in the GSSG/GSH ratio, the activity of the enzyme GSH-disulfide reductase is induced, using NADPH as a cofactor [132]. NO can inhibit GSH reductase. This relationship is not well understood, since NADPH is consumed both for NO synthesis and for GSSG reduction. There are likely as yet unidentified regulations in the glutathione and NO metabolic pathways.
CONCLUSION
For a long time, the effect of FA on the biosystem was considered from the point of view of its high toxicity. The toxicity of FA is due to its interaction with DNA (genotoxicity) and with proteins, with the formation of carbonyl derivatives. On the other hand, FA not only induces apoptosis, but also affects the proliferation of normal and cancer cells and the differentiation of stem cells. This bidirectional action is due to its concentrations, both exogenous and endogenous. For normal functioning, a system must exist in the cell to maintain the concentration of FA at a certain level, i.e. the reactions in which it is released and the reactions in which it is absorbed must be balanced. During cell transformation, the FA content may be increased, but this does not cause apoptosis, but mutagenesis. Apparently, in carcinogenesis, the primary change is the balance of FA content, while mutagenesis is secondary.
FA can be oxidized to formate and reduced to methanol with equal probability, which determined its regulatory role in the functioning of the biogenic carbon redox cycle; methanogenic archaea are methylotrophic archaea (rudiments). Evolution followed the line of methylotrophy, and methyl groups (later acetate) become proton donors for reduction reactions involving NADPH. The basic pathway for both methanogens and methylotrophs is the transfer of C1 compounds, which evolves in methylotrophs into the folate cycle and is coupled with basic metabolic pathways, including nucleotide synthesis and DNA methylation.
Endogenous FA metabolism is closely related to arginine metabolism. Apparently, through the formation of methylarginines, FA controls the synthesis of NO and through the formation of methionine, the synthesis of polyamines.
Can ADH5 activity be considered as FA detoxification? FA spontaneously interacts with both THF and GSH. Both of these interactions result in formate formation. Formate is oxidized to CO2, which can be considered as a factor in the acidification of the intracellular environment. At the same time, ADH5 is 20 times more intensive in reducing GSNO to form ammonia, which can be a factor in the alkalization of the environment. Is the activity of ADH5, an evolutionarily conserved enzyme with dual redox action - oxidation of HMGSH to formate and reduction of GSNO to ammonia - fundamental to the regulation of intracellular pH? The questions remain open.
In this regard, endogenous FA metabolism can be considered from the point of view of coupling of the main metabolic pathways, regulation of the redox system functioning and intracellular pH. The scope of this review does not allow considering coupling of FA with other metabolic pathways, in particular, the participation of serine in the regulation of pyruvate kinase activity and others.
We believe that further work expanding the systemic description of these coupled regulatory pathways will shed light on the interaction of processes that determine cell fate and its functional modes.
By using data on the kinetics of biochemical reactions, including non-enzymatic ones, and the factors of their regulation, including pH, FA, NO, and taking into account the expression of enzymes, it will be possible to construct a systemic biological model of the cell, which will allow us to obtain a description of the metabolic regimes of the cell and link them with the processes of proliferation, differentiation and apoptosis.
Conflict of Interest
Authors Laskaviy VN and Polyanina TI are founders and employees of SARBIOTEKH, which is developing a formaldehyde-based therapeutic product. This employment represents a potential conflict of interest. All other authors declare no competing interests.
Author Contributions
Tereshina EV was responsible for the conceptualization and methodology of the study, performed the formal analysis, and prepared the original draft of the manuscript. Formal analysis and critical input during manuscript revision were also provided by Alekseev AA, who was responsible for review and editing. Alekseev AA, Polyanina TI, Laskaviy VN and Sukhikh GT contributed to the discussion and interpretation of the results. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
The authors thank scientific researcher Smirnova EA for her participation in insightful discussions. Her contribution was valuable in shaping the interpretation of the findings.
REFERENCES
- Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008; 6: 579-591.
- Ward N, Larsen Ø, Sakwa J, Bruseth L, Khouri H, Durkin AS, et al. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol. 2004; 2: e303.
- Kisliuk RL. Studies on the mechanism of formaldehyde incorporation into serine. J Biol Chem. 1957; 227: 805-814.
- Tyihák E, Albert L, Németh ZI, Kátay G, Király-Véghely Z, Szende B. Formaldehyde cycle and the natural formaldehyde generators and capturers. Acta Biol Hung. 1998; 49: 225-238.
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004; 119: 941-953.
- Kubicek S, Jenuwein T. A crack in histone lysine methylation. Cell. 2004; 119: 903-906.
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature Reviews Cancer. 2013; 13: 572-583.
- Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, et al. Glycine Decarboxylase Activity Drives Non-Small Cell Lung Cancer Tumor-Initiating Cells and Tumorigenesis. Cell. 2012; 148: 259-272.
- Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2008; 84: 246-263.
- Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, et al. Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature. 2017; 548: 549-554.
- Xu Y, Meng H, Ren J, Zeng AP. Formaldehyde formation in the glycine cleavage system and its use for an aldolase-based biosynthesis of 1,3-prodanediol. J Biol Eng. 2020; 14: 15.
- Jägerstad M, Jastrebova J. 5,10-Methylene-tetrahydrofolate dissociates into tetrahydrofolate and formaldehyde at physiological pH and acidic pH, typical conditions used during sample extraction and LC-MS/MS analysis of biological samples. Biomed Chromatogr. 2014; 28: 1041-1042.
- Chen X, Chothia SY, Basran J, Hopkinson RJ. Formaldehyde regulates tetrahydrofolate stability and thymidylate synthase catalysis. Chem Commun. 2021; 57: 5778-5781.
- Tereshina EV, Laskavy VN, Ivanenko SI. Four Components of the Conjugated Redox System in Organisms: Carbon, Nitrogen, Sulfur, Oxygen. Biochem. 2015; 80: 1186-1200.
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014; 10: 298-302.
- Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metabolism. 2017; 25: 27-42.
- Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011; 108: 15163-15168.
- Stover P, Schirch V. 5-Formyltetrahydrofolate polyglutamates are slow tight binding inhibitors of serine hydroxymethyltransferase. J Biol Chem. 1991; 266: 1543-1550.
- Stover P, Schirch V. The metabolic role of leucovorin. Trends Biochem Sci. 1993; 18: 102-106.
- Paukert JL, Williams GR, Rabinowitz JC. Formyl-methenyl- methylenetetrahydrofolate synthetase (combined); correlation of enzymic activities with limited proteolytic degradation of the protein from yeast. Biochem Biophys Res Commun. 1977; 77: 147-154.
- Christensen KE, MacKenzie RE. Chapter 14 Mitochondrial Methylenetetrahydrofolate Dehydrogenase, Methenyltetrahydrofolate Cyclohydrolase,and Formyltetrahydrofolate Synthetases. Folic Acid and Folates. 2008; 393-410.
- Zhu Z, Leung GKK. More Than a Metabolic Enzyme: MTHFD2 as a Novel Target for Anticancer Therapy? Front Oncol. 2020; 10: 658.
- Nilsson R, Nicolaidou V, Koufaris C. Mitochondrial MTHFD isozymes display distinct expression, regulation, and association with cancer. Gene. 2019; 716: 144032.
- Patel H, Pietro ED, MacKenzie RE. Mammalian fibroblasts lacking mitochondrial NAD+-dependent methylenetetrahydrofolate dehydrogenase-cyclohydrolase are glycine auxotrophs. J Biol Chem. 2003; 278: 19436-19441.
- Christensen KE, Patel H, Kuzmanov U, Mejia NR, MacKenzie RE. Disruption of the mthfd1 gene reveals a monofunctional 10-formyltetrahydrofolate synthetase in mammalian mitochondria. J Biol Chem. 2005; 280: 7597-7602.
- Pike ST, Rajendra R, Artzt K, Appling DR. Mitochondrial C1- Tetrahydrofolate Synthase (MTHFD1L) Supports the Flow of Mitochondrial One-carbon Units into the Methyl Cycle in Embryos. J Biol Chem. 2010; 285: 4612-4620.
- Barlowe CK, Appling DR. In vitro evidence for the involvement of mitochondrial folate metabolism in the supply of cytoplasmic one- carbon units. Biofactors. 1988; 1: 171-176.
- Krupenko SA, Oleinik NV. 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 2002; 13: 227-236.
- Khan QA, Pediaditakis P, Malakhau Y, Esmaeilniakooshkghazi A, Ashkavand Z, Sereda V, et al. CHIP E3 ligase mediates proteasomal degradation of the proliferation regulatory protein ALDH1L1 during the transition of NIH3T3 fibroblasts from G0/G1 to S-phase. PLoS One. 2018; 13: e0199699.
- Krupenko SA. FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem Biol Interact. 2009; 178: 84-93.
- Cook RJ, Wagner C. Purification and partial characterization of rat liver folate binding protein: cytosol I. Biochemistry. 1982; 321: 4427- 4434.
- Strickland KC, Krupenko NI, Dubard ME, Hu CJ, Tsybovsky Y, Krupenko SA. Enzymatic properties of ALDH1L2, a mitochondrial 10-formyltetrahydrofolate dehydrogenase. Chemico-Biological Interactions. 2011; 191: 129-136.
- Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, Teng X, et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016; 24: 640-641.
- Krupenko NI, Dubard ME, Strickland KC, Moxley KM, Oleinik NV, Krupenko SA. ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2010; 285: 23056-23063.
- Krupenko SA, Krupenko NI. ALDH1L1 and ALDH1L2 Folate Regulatory Enzymes in Cancer. Adv Exp Med Biol. 2018; 1032: 127- 143.
- Bae S, Chon J, Field MS, Stover PJ. Alcohol Dehydrogenase 5 Is a Source of Formate for De Novo Purine Biosynthesis in HepG2 Cells. J Nutr. 2017; 147: 499-505.
- Hopkinson RJ, Schofield CJ. Deciphering Functions of Intracellular Formaldehyde: Linking Cancer and Aldehyde Metabolism. Biochem. 2018; 57: 904-906.
- Brunsdon H, Brombin A, Peterson S, Postlethwait JH, Patton EE. Aldh2 is a lineage-specific metabolic gatekeeper in melanocyte stem cells. Development. 2022; 149: dev200277.
- Sanghani PC, Stone CL, Ray BD, Pindel EV, Hurley TD, Bosron WF. Kinetic mechanism of human glutathione-dependent formaldehyde dehydrogenase. Biochemistry. 2000; 39: 10720-10729.
- Uotila L, Koivusalo M. S-Formylglutathione hydrolase. Methods Enzymol. 1981; 77: 320-325.
- Hopkinson RJ, Barlow PS, Schofield CJ, Claridge TDW. Studies on the reaction of glutathione and formaldehyde using NMR. Org Biomol Chem. 2010; 8: 4915-4920.
- Umansky C, Morellato AE, Rieckher M, Scheidegger MA, Martinefski MR, Fernández GA, et al. Endogenous formaldehyde scavenges cellular glutathione resulting in redox disruption and cytotoxicity. Nat Commun. 2022; 13: 745.
- He H, Noor E, Ramos-Parra PA, García-Valencia LE, Patterson JA, de la Garza RID, et al. In Vivo Rate of Formaldehyde Condensation with Tetrahydrofolate. Metabolites. 2020; 10: 65.
- Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA- repair pathway. Nat Struct Mol Biol. 2011; 18: 1432-1434.
- Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell. 2015; 60: 177- 188.
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004; 116: 617-628.
- Wang R-S, Nakajima T, Kawamoto T, Honma T. Effects of Aldehyde Dehydrogenase-2 Genetic Polymorphisms on Metabolism of Structurally Different Aldehydes in Human Liver. Drug Metabolism and Disposition. 2002; 30: 69-73.
- Jacobson MK, Bernofsky C. Mitochondrial acetaldehyde dehydrogenase from Saccharomyces cerevisiae. Biochim Biophys Acta. 1974; 350: 277-291.
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, et al. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Current Biol. 1997; 7: 427-439.
- Dingler FA, Wang M, Mu A, Millington CL, Oberbeck N, Watcham S, et al. Two Aldehyde Clearance Systems Are Essential to Prevent Lethal Formaldehyde Accumulation in Mice and Humans. Mol Cell. 2020; 80: 996-1012.e9.
- Nannelli G, Terzuoli E, Giorgio V, Donnini S, Lupetti P, Giachetti A, et al. ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid Med Cell Longev. 2018; 2018: 9765027.
- Tyihák E, Bocsi J, Timár F, Rácz G, Szende B. Formaldehyde promotes and inhibits the proliferation of cultured tumour and endothelial cells. Cell Prolif. 2001; 34: 135-141.
- Szende B, Tyihák E. Effect of formaldehyde on cell proliferation and death. Cell Biol Int. 2010; 34: 1273-1282.
- Teng S, Beard K, Pourahmad J, Moridani M, Easson E, Poon R, et al. The formaldehyde metabolic detoxification enzyme systems and molecular cytotoxic mechanism in isolated rat hepatocytes. Chem Biol Interact. 2001; 130-132: 285-296.
- Anguera MC, Field MS, Perry C, Ghandour H, Chiang E-P, Selhub J, et al. Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2006; 281: 18335-18342.
- Machlin LJ. Handbook of Vitamins: Nutritional, Biochemical, and Clinical Aspects. Marcel Dekker Incorporated; 1984.
- Pilch B, Mann M. Large-scale and high-confidence proteomic analysis of human seminal plasma. Genome Biol. 2006; 7: R40.
- Kutzbach C, Stokstad ELR. Feedback inhibition of methylene- tetrahydrofolate reductase in rat liver by S-adenosylmethionine. Biochimica et Biophysica Acta (BBA) - Enzymol. 1967; 217-220.
- Jencks DA, Mathews RG. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J Biol Chem. 1987; 262: 2485-2493.
- Cook RJ, Wagner C. Glycine N-methyltransferase is a folate binding protein of rat liver cytosol. Proc Natl Acad Sci. 1984; 3631-3634.
- Luka Z. Chapter 11 Methyltetrahydrofolate in Folate-Binding Protein Glycine N-Methyltransferase. Folic Acid and Folates. 2008; 325-345.
- Wang YC, Chen YM, Lin YJ, Liu SP, Chiang EPI. GNMT expression increases hepatic folate contents and folate-dependent methionine synthase-mediated homocysteine remethylation. Mol Med. 2011; 17: 486-494.
- DebRoy S, Kramarenko II, Ghose S, Oleinik NV, Krupenko SA, Krupenko NI. A novel tumor suppressor function of glycine N-methyltransferase is independent of its catalytic activity but requires nuclear localization. PLoS One. 2013; 8: e70062.
- Yen CH, Lu YC, Li CH, Lee CM, Chen CY, Cheng MY, et al. Functional characterization of glycine N-methyltransferase and its interactive protein DEPDC6/DEPTOR in hepatocellular carcinoma. Mol Med. 2012;18: 286-296.
- Huszti Z, Tyihák E. Formation of formaldehyde from S-adenosyl- L-[methyl-3H]methionine during enzymic transmethylation of histamine. FEBS Lett. 1986; 209: 362-366.
- Pervin S, Singh R, Hernandez E, Wu G, Chaudhuri G. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007; 67: 289-299.
- Hümmer J, Kraus S, Brändle K, Lee-Thedieck C. Nitric Oxide in the Control of the Proliferation and Differentiation of Human Hematopoietic Stem and Progenitor Cells. Front Cell Dev Biol. 2020; 8: 610369.
- Tejedo JR, Tapia-Limonchi R, Mora-Castilla S, Cahuana GM, Hmadcha A, Martin F, et al. Low concentrations of nitric oxide delay the differentiation of embryonic stem cells and promote their survival. Cell Death Dis. 2010; 1: e80.
- Wong JC, Fiscus RR. Essential roles of the nitric oxide (no)/cGMP/ protein kinase G type-Iα (PKG-Iα) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Iα autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. J Cell Biochem. 2011; 112: 829-839.
- Frank S, Kämpfer H, Podda M, Kaufmann R, Pfeilschifter J. Identification of copper/zinc superoxide dismutase as a nitric oxide- regulated gene in human (HaCaT) keratinocytes: implications for keratinocyte proliferation. Biochem J. 2000; 346: 719-728.
- Melendez JA, Melathe RP, Rodriguez AM, Mazurkiewicz JE, Davies KJ. Nitric oxide enhances the manganese superoxide dismutase- dependent suppression of proliferation in HT-1080 fibrosarcoma cells. Cell Growth Differ. 1999; 10: 655-664.
- Pignatti C, Tantini B, Stefanelli C, Giordano E, Bonavita F, Cl C, et al. Nitric oxide mediates either proliferation or cell death in cardiomyocytes. Involvement of polyamines. Amino Acids. 1999; 181-190.
- Poderoso JJ, Lisdero C, Schöpfer F, Riobó N, Carreras MC, Cadenas E, et al. The regulation of mitochondrial oxygen uptake by redoxreactions involving nitric oxide and ubiquinol. J Biol Chem. 1999; 274: 37709-37716.
- Orsi A, Beltrán B, Clementi E, Hallén K, Feelisch M, Moncada S. Continuous exposure to high concentrations of nitric oxide leads to persistent inhibition of oxygen consumption by J774 cells as well as extraction of oxygen by the extracellular medium. Biochem J. 2000; 346: 407–412.
- Baker MS, Gebicki JM. The effect of pH on the conversion of superoxide to hydroxyl free radicals. Arch Biochem Biophys. 1984; 258-264.
- Stuehr DJ, Kwon NS, Nathan CF. FAD and GSH participate in macrophage synthesis of nitric oxide. Biochem Biophys Res Commun. 1990; 168: 558-565.
- Messina JP, Lawrence DA. Cell cycle progression of glutathione- depleted human peripheral blood mononuclear cells is inhibited at S phase. J Immunol. 1989; 143: 1974-1981.
- Gamcsik MP, Kasibhatla MS, Teeter SD, Colvin OM. Glutathione levels in human tumors. Biomarkers. 2012; 17: 671-691.
- Carretero J, Obrador E, Anasagasti MJ, Martin JJ, Vidal-Vanaclocha F, Estrela JM. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin Exp Metastasis. 1999; 17: 567-574.
- Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001; 410: 490-494.
- Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003; 9: 160-168.
- Tillmann AT, Strijbis K, Cameron G, Radmaneshfar E, Thiel M, Munro CA, et al. Contribution of Fdh3 and Glr1 to Glutathione Redox State, Stress Adaptation and Virulence in Candida albicans. PLoS One. 2015;10: e0126940.
- Hedberg JJ, Griffiths WJ, Nilsson SJF, Höög J-O. Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analysed by electrospray mass spectrometry. Eur J Biochem. 2003; 270: 1249-1256.
- Barnett SD, Buxton ILO. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit Rev Biochem Mol Biol. 2017; 52: 340-354.
- Lamb J, Wheatley DN. Single amino acid (arginine) deprivation induces G1 arrest associated with inhibition of cdk4 expression in cultured human diploid fibroblasts. Exp Cell Res. 2000; 255: 238-249.
- Zhang WY, Takiguchi M, Koshiyama Y, Gotoh T, Nagasaki A, Iwase K, et al. Expression of citrulline-nitric oxide cycle in lipopolysaccharide and cytokine-stimulated rat astroglioma C6 cells. Brain Res. 1999; 849: 78-84.
- Nussler AK, Liu ZZ, Hatakeyama K, Geller DA, Billiar TR, Morris SM Jr. A cohort of supporting metabolic enzymes is coinduced with nitric oxide synthase in human tumor cell lines. Cancer Lett. 1996; 103: 79- 84.
- Keshet R, Erez A. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. Dis Model Mech. 2018; 11.
- Tyihák E, Szende B, Lapis K. Biological significance of methylated derivatives of lysine and arginine. Life Sci. 1977; 20: 385-392.
- Peng C, Wong CC. The story of protein arginine methylation: characterization, regulation, and function. Expert Rev Proteomics. 2017; 14: 157-170.
- Petrossian TC, Clarke SG. Uncovering the human methyltransferasome.Mol Cell Proteomics. 2011; 10: M110.000976.
- Fulton MD, Brown T, Zheng YG. The Biological Axis of Protein Arginine Methylation and Asymmetric Dimethylarginine. Int J Mol Sci. 2019; 20.
- Bedford MT. Arginine methylation at a glance. J Cell Sci. 2007; 120: 4243-4246.
- Strobel J, Müller F, Zolk O, Endreß B, König J, Fromm MF, et al. Transport of asymmetric dimethylarginine (ADMA) by cationic amino acid transporter 2 (CAT2), organic cation transporter 2 (OCT2) and multidrug and toxin extrusion protein 1 (MATE1). Amino Acids. 2013; 45: 989-1002.
- Davids M, Teerlink T. Plasma concentrations of arginine and asymmetric dimethylarginine do not reflect their intracellular concentrations in peripheral blood mononuclear cells. Metabolism. 2013; 62: 1455-1461.
- Teerlink T, Luo Z, Palm F, Wilcox CS. Cellular ADMA: regulation and action. Pharmacol Res. 2009; 60: 448-460.
- Zakrzewicz D, Eickelberg O. From arginine methylation to ADMA: a novel mechanism with therapeutic potential in chronic lung diseases. BMC Pulm Med. 2009; 9: 5.
- Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal. 2016; 9: rs9.
- Fuhrmann J, Clancy KW, Thompson PR. Chemical biology of protein arginine modifications in epigenetic regulation. Chem Rev. 2015; 115: 5413-5461.
- Closs EI, Basha FZ, Habermeier A, Förstermann U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide. 1997; 1: 65-73.
- Kakimoto Y, Akazawa S. Isolation and identification of N-G,N-G- and N-G,N’-G-dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta- hydroxylysine from human urine. J Biol Chem. 1970; 245: 5751- 5758.
- Yu PH, Wright S, Fan EH, Lun Z-R, Gubisne-Harberle D. Physiological and pathological implications of semicarbazide-sensitive amine oxidase. Biochim Biophys Acta. 2003; 1647: 193-199.
- Cardounel AJ, Xia Y, Zweier JL. Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetrahydrobiopterin. J Biol Chem. 2005; 280: 7540-7549.
- Druhan LJ, Forbes SP, Pope AJ, Chen C-A, Zweier JL, Cardounel AJ. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry. 2008; 47: 7256-7263.
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990; 87: 1620-1624.
- Trézl L, Hullán L, Szarvas T, Csiba A, Szende B. Determination of endogenous formaldehyde in plants (fruits) bound to L-arginine and its relation to the folate cycle, photosynthesis and apoptosis. Acta Biol Hung. 1998; 49: 253-263.
- Hullán L, Trézl L, Szarvas T, Csiba A. The hydrazine derivative aminoguanidine inhibits the reaction of tetrahydrofolic acid with hydroxymethylarginine biomolecule. Acta Biol Hung. 1998; 49: 265-273.
- Szende B, Tyihák E, Trézl L, Szöke E, László I, Kátay G, et al. Formaldehyde generators and capturers as influencing factors of mitotic and apoptotic processes. Acta Biol Hung. 1998; 49: 323-329.
- Szende B, Tyihák E, Trézl L. Role of arginine and its methylated derivatives in cancer biology and treatment. Cancer Cell Int. 2001; 1: 3.
- Leu SY, Wang SR. Clinical significance of arginase in colorectal cancer. Cancer. 1992; 70: 733-736.
- Straus B, Cepelak I, Festa G. Arginase, a new marker of mammary carcinoma. Clin Chim Acta. 1992; 210: 5-12.
- Pegg AE. Spermidine/spermine-N(1)-acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab. 2008; 294: E995-1010.
- Casero RA, Pegg AE. Polyamine catabolism and disease. Biochem J. 2009; 421: 323-338.
- Bachrach U, Wang YC, Tabib A. Polyamines: new cues in cellular signal transduction. News Physiol Sci. 2001; 16: 106-109.
- Childs AC, Mehta DJ, Gerner EW. Polyamine-dependent gene expression. Cell Mol Life Sci. 2003; 60: 1394-1406.
- Bardocz S, White A. Polyamines in Health and Nutrition. Springer Science & Business Media; 1999.
- Seiler N, Raul F. Polyamines and apoptosis. J Cell Mol Med. 2005; 9: 623-642.
- Roholt OA Jr, Greenberg DM. Liver arginase. IV. Effect of pH on kinetics of manganese-activated enzyme. Arch Biochem Biophys. 1956; 62: 454-470.
- Daghigh F, Fukuto JM, Ash DE. Inhibition of rat liver arginase by an intermediate in NO biosynthesis, NG-hydroxy-L-arginine: implications for the regulation of nitric oxide biosynthesis by arginase. Biochem Biophys Res Commun. 1994; 202: 174-180.
- Singh R, Pervin S, Karimi A, Cederbaum S, Chaudhuri G. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L- arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000; 60: 3305-3312.
- Buga GM, Wei LH, Bauer PM, Fukuto JM, Ignarro LJ. NG-hydroxy-L- arginine and nitric oxide inhibit Caco-2 tumor cell proliferation by distinct mechanisms. Am J Physiol. 1998; 275: R1256-R1264.
- Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res. 2007; 101: 692-702.
- Mössner J, Hammermann R, Racké K. Concomitant down-regulation of L-arginine transport and nitric oxide (NO) synthesis in rat alveolar macrophages by the polyamine spermine. Pulm Pharmacol Ther. 2001; 14: 297-305.
- Bauer PM, Buga GM, Fukuto JM, Pegg AE, Ignarro LJ. Nitric oxide inhibits ornithine decarboxylase via S-nitrosylation of cysteine 360 in the active site of the enzyme. J Biol Chem. 2001; 276: 34458- 34464.
- Casero RA Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007; 6: 373-390.
- Shantz LM, Levin VA. Regulation of ornithine decarboxylase during oncogenic transformation: mechanisms and therapeutic potential. Amino Acids. 2007; 33: 213-223.
- Luk GD, Moshier JA, Ehrinpreis MN. Ornithine decarboxylase as a marker for colorectal polyps and cancer. Prog Clin Biol Res. 1988; 279: 227-239.
- Upp JR Jr, Saydjari R, Townsend CM Jr, Singh P, Barranco SC, Thompson JC. Polyamine levels and gastrin receptors in colon cancers. Ann Surg. 1988; 207: 662-669.
- Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U S A. 1993; 90: 7804-7808.
- Tobias KE, Shor J, Kahana C. c-Myc and Max transregulate the mouse ornithine decarboxylase promoter through interaction with two downstream CACGTG motifs. Oncogene. 1995; 11: 1721-1727.
- Owen JB, Butterfield DA. Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol. 2010; 648: 269-277.
- López-Mirabal HR, Winther JR. Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta. 2008; 1783: 629-640.









































































































































{kind=link}